Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roland Kuiper | + 2303 word(s) | 2303 | 2021-05-18 04:35:04 | | | |
| 2 | Bruce Ren | -21 word(s) | 2282 | 2021-05-25 06:28:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kuiper, R. Antigen Targeted Therapy for LS. Encyclopedia. Available online: https://encyclopedia.pub/entry/10001 (accessed on 07 February 2026).
Kuiper R. Antigen Targeted Therapy for LS. Encyclopedia. Available at: https://encyclopedia.pub/entry/10001. Accessed February 07, 2026.
Kuiper, Roland. "Antigen Targeted Therapy for LS" Encyclopedia, https://encyclopedia.pub/entry/10001 (accessed February 07, 2026).
Kuiper, R. (2021, May 24). Antigen Targeted Therapy for LS. In Encyclopedia. https://encyclopedia.pub/entry/10001
Kuiper, Roland. "Antigen Targeted Therapy for LS." Encyclopedia. Web. 24 May, 2021.
Copy Citation
Lynch syndrome (LS) and constitutional mismatch repair deficiency (CMMRD) are hereditary disorders which significantly increase a person’s risk of developing a variety of cancers such as colorectal, endometrial, brain and, for CMMRD also, haematological malignancies. This increased cancer risk is due to inherited mutations in specific types of DNA repair genes, which hampers repair of mispaired or damaged bases during DNA replication. As a consequence, somatic mutations rapidly accumulate and typically include insertions and deletions (indels) in microsatellites that potentially can give rise to neoantigens. These neoantigens open up avenues for neoantigen-targeting immune therapies.
Lynch Syndrome
hereditary cancer
CMMRD
neoantigen
colorectal cancer
mismatch repair deficiency
targeted therapy
1. Introduction
Lynch Syndrome (LS) is an autosomal dominantly inherited disorder resulting from monoallelic germline aberrations in genes that are involved in DNA mismatch repair (MMR) machinery [1]. The four MMR genes that are implicated in the disorder are MLH1, MSH2, MSH6 and PMS2 [2]. Patients with LS inherit a pathogenic germline variant in only one allele while the remaining wild type allele is somatically inactivated by point mutations, loss of heterozygosity or epigenetic silencing due to promoter hypermethylation [3][4]. In 1999, two reports described the phenotype within LS families including children who carried homozygous germline mutations in the MLH1 gene. The children in both families displayed haematological malignancies in early childhood and clinical features that were previously known from neurofibromatosis type 1 [5][6]. Since then, individuals who inherit bi-allelic germline mutations in one of the MMR genes have been identified to suffer from constitutional mismatch repair deficiency (CMMRD). This rare syndrome is inherited recessively with homozygous or compound heterozygous germline mutations in the DNA MMR genes, most commonly PMS2 and MSH6 [7].
LS increases a person’s risk of colorectal cancer (CRC) by 40–80% and endometrial cancer by 15–60% [8]. Individuals with LS are also more prone to a variety of cancers among which are urothelial (0.4–20%), ovarian cancers (4–12%), gastric cancers (<10%), brain tumours and also cancers of the biliary tract [9][10][11][12]. Similarly, CMMRD patients have an increased risk of developing CRC in adolescence or young adulthood. In patients with CMMRD 50% develop malignant brain tumours while 40% develop cancers of the digestive tract [13]. The risk of developing haematological malignancies is as high as 30% [13]. In fact, the penetrance of cancers in CMMRD is one of the highest among childhood cancer syndromes, and it is extremely uncommon for a patient not to have developed cancer by the third decade [14]. The increased cancer risk in LS patients stems from the loss of the second functional MMR allele which results in accumulation of somatic mutations leading to carcinogenesis [15]. In contrast, tumorigenesis in CMMRD patients does not depend on second hit mutations since the biallelic loss of MMR functioning itself renders the cells unable to repair damaged DNA and hence lose genomic integrity [16]. Figure 1 summarises the key features of LS and CMMRD.
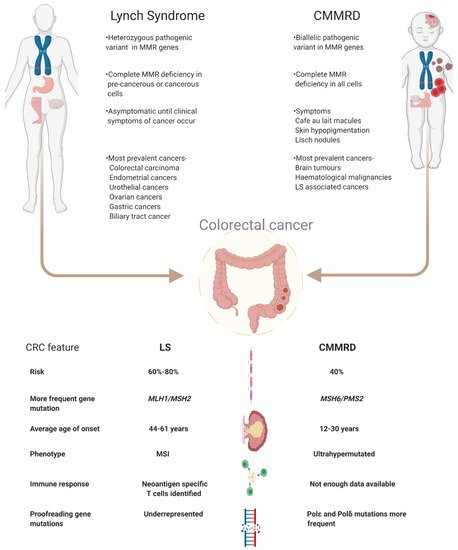
Figure 1. Schematic view on the differences between LS and CMMRD with a focus on colorectal cancer. Abbreviations used- LS: Lynch Syndrome, CMMRD: Constitutional mismatch repair deficiency syndrome, MMR: mismatch repair, CRC: colorectal cancer, MSS: microsatellite stable, MSI: microsatellite instable.
The accumulation of somatic mutations and genomic instability, especially in mutation prone regions, e.g., regions of repetitive nucleotide sequences, results in non-synonymous mutations. These mutations give rise to proteins with altered amino acid sequences called frameshift peptides (FSPs) that can give rise to neoantigens [17][18]. Neoantigens make an attractive target for immunotherapies since they have not been subjected to central and thymic tolerance and are solely expressed by tumour cells [19]. Tumours with high mutational burden such as those in LS and CMMRD patients are more likely to give rise to neoantigens and hence provide more opportunities for targeted therapies [18]. Despite the presence of technological facilities that help with the efficient identification of such neoantigens, therapies henceforth developed are still in nascent stages when compared to neoantigen targeting therapies in melanoma which have shown tumour regression in patients [20]. This demands further probe into the aspects that are impairing a successful neoantigen targeting regimen in LS and CMMRD.
2. Mismatch Repair Deficiency and Microsatellite Instability
DNA damage can occur endogenously through metabolic processes inside a cell and through exogenous processes like environmental agents. The repair pathways involved in the repair of damaged DNA are broadly classified as base excision repair, homologous recombination, non-homologous end joining, nucleotide excision repair and MMR [21][22]. The role of the DNA MMR system is to maintain genomic integrity through base pair and small insertion-deletion (indel) corrections that are erroneously generated during DNA replication [23]. The most important components of the DNA MMR system are the MutS and MutL complexes. In its functional state, MutSα, consisting of MSH2 and MSH6 proteins, recognises single base indels. Functional MutSβ, consisting of MSH2 and MSH3 proteins, recognises indels consisting of 2–8 nucleotides. MutLα, consisting of MLH1 and PMS2, or MutLβ, consisting of MLH1 and PMS1, binds together with the MutS complex as a heterodimer along with replicative factors to initiate repair of the mismatched nucleotides. Since MSH2 and MLH1 are the proteins shared by both components of their respective MutS and MutL complexes, a mutation in the respective genes will completely retard all MMR activity whereas a mutation in PMS2 or MSH6 genes will reduce MMR activity towards single nucleotide indels only [24]. Tumours arise from clonal expansion of cells that have inactivation of both alleles of a MMR gene that can either be somatic or of germline origin.
Indels commonly occur in regions of repetitive nucleotide sequences called microsatellites, where the template and the primer strands are prone to slippage (i.e., dissociation and re-annealing) during replication. Such mismatches are not repaired in MMR deficient cells, resulting in an incorrect number of repeat units between the template and newly synthesised strand. The microsatellite alterations can lead to a shift in the translational reading frame and therefore generation of FSPs [25]. This genetic alteration is termed microsatellite instability (MSI) and is a characteristic of LS-associated cancers. The process is briefly summarised in Figure 2. MSI is not specific for LS and CMMRD, approximately 15% of sporadic colorectal cancers also demonstrate MSI that most often originates by hypermethylation of the MLH1 promotor and somatic bi-allelic inactivation of MMR genes [26][27][28]. While all microsatellites have an equal chance for mutations, differences in their mutation frequency can occur due the length of the repeat and the nature of the relevant nucleotide sequence.
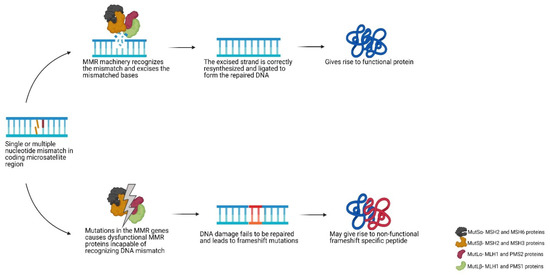
Figure 2. Schematic view of the DNA mismatch repair pathway. Abbreviations used- MMR: mismatch repair.
3. Clinical Management of LS and CMMRD
Since LS patients are at risk of early onset CRC, regular colonoscopy surveillance starting from age 20–25, is essential to diagnose early lesions with the intent to prevent development of CRC. For LS patients, regular coloscopy is quite a burden and does not prevent the formation of new lesions, pointing to the need for other preventive measures [29]. There is evidence of reduced CRC risk in LS patients and sporadic MMR gene mutation carriers who took 600 mg/day aspirin for at least 2 years [30]. However, there are concerns regarding the risk of bleeding events in young patients [31]. Since a subset of chemotherapeutics rely on a functional MMR system to induce tumour damage, the efficacy of such drugs in MMR-deficient tumours such as in LS or CMMRD has been poor [32]. In fact worse prognosis was seen for stage II MSI high (MSI-H) colon cancer patients in a randomised trial administering adjuvant based 5-Flurouracil chemotherapy (5-FU) as compared to microsatellite stable (MSS) tumours, as a result of a lesser effectiveness of 5-FU in these MSI-H cancers [33]. The resistance of MMR deficient cells to drugs such as temozolomide, an alkylating agent used to treat glioblastoma multiforme (GBM), can lead to a greater risk of developing second primary tumours in CMMRD patients because of the accumulation of unrepaired mutations [7]. MMR-deficient cells are also more resistant to cisplatin treatment in comparison to MMR-proficient cells [34][35].
MSI-H cancers have a higher density of infiltrating lymphocytes compared to MSS cancers, which has been demonstrated to correlate with a better prognosis [36][37]. This observation highlights the potential of immunotherapy. In fact, LS-associated cancers have more pronounced local immune responses as compared to sporadic MSI-H cancers [38]. However, increased infiltration is counteracted by increased checkpoint protein expression which is an important mechanism by which the tumour microenvironment inhibits immune responses. By chronically expressing checkpoint receptors such as CTLA-4, PD-1, TIM-3, LAG-3 and more, T-cells become functionally exhausted and dysfunctional [39]. The upside of this checkpoint protein overexpression phenotype is the strong efficacy of immune checkpoint inhibitor therapies that have shown positive outcomes in MMR-deficient tumours across a range of malignancies [40]. This outcome has already led to FDA approval of the PD-1 antibody pembrolizumab for the treatment of refractory dMMR/MSI-H solid malignancies, and the PD-1 antibody nivolumab with or without the CTLA-4 antibody ipilimumab for the treatment of dMMR/MSI-H CRC after 5-FU treatment [41]. In a trial combining nivolumab and ipilimumab (NCT02060188), a disease control rate of 79% for >12 weeks was reported irrespective of clinical LS history [42]. Phase II studies have shown the safety and durable efficacy of nivolumab in patients with advanced MMR-deficient CRC (NCT01876511) where an immune-related progression free survival rate was seen for 78% of patients with MMR-deficient cancers as compared to the 11% of patients that had MMR-proficient cancers. However, it is important to note that while objective responses were seen in 100% of all non-LS-associated MMR-deficient cancers, only 27% (3 of 11 patients) of LS-associated cancers showed an objective response [40]. In a case study of a patient with LS-associated metastatic CRC, pembolizumab treatment reduced the metabolic activity of the cancerous lesions and improved symptoms [43]. Additionally, another case report from a LS patient with pancreatic adenocarcinoma and metastatic liver disease showed excellent clinical response with regards to liver lesion shrinkage after only one cycle of pembrolizumab treatment [44]. Nivolumab has also been reported to have significant efficacy at inducing an anti-tumour response and prolonging survival for two patients with CMMRD recurrent glioblastoma [45]. In another young child with CMMRD-associated GBM, nivolumab therapy showed a 60% reduction in tumour size and improved symptoms [46]. These groundbreaking results have amplified interest in the potential use of checkpoint inhibitors in combination with other therapeutics in the treatment of MMR-deficient cancers in LS and CMMRD.
4. Targeting Neoantigens in LS and CMMRD
Tumours that arise due to LS or CMMRD are highly mutated compared to their MMR-proficient countertypes. For instance, paediatric glioblastomas in CMMRD patients exhibit an ultra-high number of nonsynonymous mutations (≥250 mut/Mb) which contrasts the low frequency of nonsynonymous mutations (<1 mut/Mb) seen in the majority of glioblastomas [47][48].
The high tumour mutational burden (TMB) generated because of the MMR defect can result in neoantigen formation. These neoantigens are formed when indels result in a frameshift of the amino acid sequence in the C-terminal of the protein producing an FSP, that acts as a substrate for antigen processing and presentation via the major histocompatibility complex (MHC) class I and II molecules [49]. Once presented on the cell surface, they are referred to as neoantigens and could act as targets for tumour-infiltrating CD4+ helper T lymphocytes and CD8+ cytotoxic T lymphocytes (CTLs). Various clinical trials employing neoantigen based therapies for CRC are already ongoing [50].
Several studies have identified neoantigens in LS patients that are highly immunogenic, for example TGFβRII, CASP5, TAF1B, HT001 and OGT [51][52]. More importantly, CTLs specific to these neoantigens have also been detected in LS patients. CTLs directed against TGFβRII and CASP5 neoantigens are capable of lysing MSI-H colon carcinoma cells as shown in in-vitro assays [52][53][54]. Some of these mutations are found to be shared between LS and non-LS MSI-H CRCs hence calling attention to neoantigen-targeting therapies encompassing more patient groups [55]. Moreover CTLs specific for a wide range of neoantigens have been found to be induced already in patients that have not yet developed a cancerous lesion i.e., in healthy LS carriers [52]. This points towards the strong immune surveillance mechanisms in LS patients whereby the immune system recognises and possibly has the potential of eradicating MMR-deficient cells even before they develop into cancer. These observations strongly argue in favour of a promising efficacy of neoantigen-targeting therapy for therapeutic and preventive purposes in LS and CMMRD with added significance for prophylactic purposes to prevent admission of chemotherapy.
Strong lymphocyte infiltration in MMR deficient cancers makes it a prime target for checkpoint inhibitor therapy, some of which have been discussed above. In addition, it opens avenues for other immunotherapies such as dendritic cell (DC) vaccination. DC vaccination may lead to the specific enhancement of immune responses against neoantigens and hence lesser toxicity as opposed to general immune activation in response to checkpoint inhibition. This is already being investigated in a DC vaccination trial in LS mutation carriers, to assess the feasibility of DC vaccination loaded with frameshift derived neoantigens associated with MSI (NCT01885702). Preliminary data show that after DC vaccination, neoantigen-specific T-cells are detectable in blood and delayed type hypersensitivity (DTH) tests, without the induction of severe adverse events [56]. Similarly, in another Phase I/II trial (NCT01461148) in LS patients, a vaccine against the neoantigens HT001, AIM2 and TAF1B has been shown to be well tolerated with no severe adverse effects in any patient, and induced humoral and cellular responses in all patients [57]. Another recent case study investigated the use of a combination of autologous DCs producing IL-12, nivolumab (anti-PD1 receptor) and radiotherapy for the treatment of a CMMRD patient that showed a complete therapeutic response [58]. Another promising immunotherapeutic approach was demonstrated preclinically, with the adoptive transfer of CTLs with an engineered TCR-directed against a FSP of the TGFβRII gene. This particular TGFβRII frameshift mutation is reported in 90% of MSI-H CRC [59]. The adoptive transfer induced significant reduction in tumour load in a xenograft mouse model [60]. However, this approach has not been tested in humans yet.
As promising as these strategies are, there are still many obstacles that need to be overcome.
References
- Hampel, H.; Hoffner, B. Lynch Syndrome and Immunotherapy. J. Adv. Pract. Oncol. 2017, 8, 7–21.
- Lynch, H.T.; Lanspa, S.; Shaw, T.; Casey, M.J.; Rendell, M.; Stacey, M.; Townley, T.; Snyder, C.; Hitchins, M.; Bailey-Wilson, J. Phenotypic and genotypic heterogeneity of Lynch syndrome: A complex diagnostic challenge. Fam. Cancer 2018, 17, 403–414.
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162.
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793.
- Ricciardone, M.D.; Tayfun, O. Advances in Brief Human MLH1 Deficiency Predisposes to Hematological Malignancy and. Cancer Res. 1999, 59, 290–293.
- Wang, Q.; Lasset, C.; Desseigne, F.; Saurin, J.-C.; Maugard, C.; Navarro, C.; Ruano, E.; Descos, L.; Trillet-Lenoir, V.; Bosset, J.-F.; et al. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum. Genet. 1999, 105, 79–85.
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.A.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.-M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J. Med. Genet. 2014, 51, 355–365.
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565.
- Bucksch, K.; Zachariae, S.; Aretz, S.; Büttner, R.; Holinski-Feder, E.; Holzapfel, S.; Hüneburg, R.; Kloor, M.; Von Knebel Doeberitz, M.; Morak, M.; et al. Cancer risks in Lynch syndrome, Lynch-like syndrome, and familial colorectal cancer type X: A prospective cohort study. BMC Cancer 2020, 20, 1–11.
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise management of ovarian cancer: On the cutting edge. J. Pers. Med. 2020, 10, 41.
- Van Der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.L.; Witjes, J.A.; Hulsbergen-Van De Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; Van Krieken, J.H.J.M.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med. Genet. 2010, 47, 464–470.
- Mankaney, G.; Macaron, C.; Burke, C.A. Refining Risk Factors for Gastric Cancer in Patients With Lynch Syndrome to Optimize Surveillance Esophagogastroduodenoscopy. Clin. Gastroenterol. Hepatol. 2020, 18, 780–782.
- Abedalthagafi, M. Constitutional mismatch repair-deficiency: Current problems and emerging therapeutic strategies. Oncotarget 2018, 9, 35458–35469.
- Lavoine, N.; Colas, C.; Muleris, M.; Bodo, S.; Duval, A.; Entz-Werle, N.; Coulet, F.; Cabaret, O.; Andreiuolo, F.; Charpy, C.; et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J. Med. Genet. 2015, 52, 770–778.
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194.
- Westdorp, H.; Kolders, S.; Hoogerbrugge, N.; de Vries, I.J.M.; Jongmans, M.C.J.; Schreibelt, G. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Lett. 2017, 403, 159–164.
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 1–12.
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 1–14.
- George, J.T.; Kessler, D.A.; Levine, H. Effects of thymic selection on T cell recognition of foreign and tumor antigenic peptides. Proc. Natl. Acad. Sci. USA 2017, 114, E7875–E7881.
- Zhou, J.; Dudley, M.E.; Rosenberg, S.A.; Robbins, P.F. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother. 2005, 28, 53–62.
- Burgess, J.T.; Rose, M.; Boucher, D.; Plowman, J.; Molloy, C.; Fisher, M.; O’Leary, C.; Richard, D.J.; O’Byrne, K.J.; Bolderson, E. The Therapeutic Potential of DNA Damage Repair Pathways and Genomic Stability in Lung Cancer. Front. Oncol. 2020, 10, 1–14.
- Boussios, S.; Moschetta, M.; Karihtala, P.; Samartzis, E.P.; Sheriff, M.; Pappas-Gogos, G.; Ozturk, M.A.; Uccello, M.; Karathanasi, A.; Tringos, M.; et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann. Transl. Med. 2020, 8, 1706.
- Colle, R.; Cohen, R.; Cochereau, D.; Duval, A.; Lascols, O.; Lopez-Trabada, D.; Afchain, P.; Trouilloud, I.; Parc, Y.; Lefevre, J.H.; et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull. Cancer 2017, 104, 42–51.
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179.
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res. 2004, 64, 3014–3021.
- Vos, J.R.; Fakkert, I.E.; Spruijt, L.; Willems, R.W.; Langenveld, S.; Mensenkamp, A.R.; Leter, E.M.; Nagtegaal, I.D.; Ligtenberg, M.J.L.; Hoogerbrugge, N. Evaluation of yield and experiences of age-related molecular investigation for heritable and nonheritable causes of mismatch repair deficient colorectal cancer to identify Lynch syndrome. Int. J. Cancer 2020, 147, 2150–2158.
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346.
- Elze, L.; Mensenkamp, A.R.; Nagtegaal, I.D.; van Zelst-Stams, W.; de Voer, R.M.; Ligtenberg, M.J.L. Somatic Nonepigenetic Mismatch Repair Gene Aberrations Underly Most Mismatch Repair-Deficient Lynch-Like Tumors. Gastroenterology 2021, 160, 1414–1416.e3.
- van Leerdam, M.E.; Roos, V.H.; van Hooft, J.E.; Balaguer, F.; Dekker, E.; Kaminski, M.F.; Latchford, A.; Neumann, H.; Ricciardiello, L.; Rupinska, M.; et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019, 51, 1082–1093.
- Umar, A. Faculty Opinions recommendation of Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Fac. Opin. 2011.
- Leenders, E.K.S.M.; Westdorp, H.; Brüggemann, R.J.; Loeffen, J.; Kratz, C.; Burn, J.; Hoogerbrugge, N.; Jongmans, M.C.J. Cancer prevention by aspirin in children with Constitutional Mismatch Repair Deficiency (CMMRD). Eur. J. Hum. Genet. 2018, 26, 1417–1423.
- Jo, W.-S.; Carethers, J.M. Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomark. 2006, 2, 51–60.
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3219–3226.
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch repair and DNA damage signalling. DNA Repair 2004, 3, 1091–1101.
- Sawant, A.; Kothandapani, A.; Zhitkovich, A.; Sobol, R.W.; Patrick, S.M. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 2015, 35, 126–136.
- Seth, S.; Ager, A.; Arends, M.J.; Frayling, I.M. Lynch syndrome—Cancer pathways, heterogeneity and immune escape. J. Pathol. 2018, 246, 129–133.
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422.
- Bohaumilitzky, L.; von Knebel Doeberitz, M.; Kloor, M.; Ahadova, A. Implications of Hereditary Origin on the Immune Phenotype of Mismatch Repair-Deficient Cancers: Systematic Literature Review. J. Clin. Med. 2020, 9, 1741.
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530.
- Diaz, L.A., Jr.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979.
- Gurjao, C.; Liu, D.; Hofree, M.; AlDubayan, S.H.; Wakiro, I.; Su, M.-J.; Felt, K.; Gjini, E.; Brais, L.K.; Rotem, A.; et al. Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair-Deficient Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 1230–1236.
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779.
- Salman, P.; Panay, S.; Fernández, R.; Mahave, M.; Soza-Ried, C. Evidence of response to pembrolizumab in a patient with lynch syndrome-related metastatic colon cancer. Onco Targets Ther. 2018, 11, 7295–7300.
- Patil, N.R.; Khan, G.N. Exceptional response to a single cycle of immunotherapy in a lynch syndrome patient with metastatic pancreatic adenocarcinoma. Am. J. Case Rep. 2020, 21, 1–6.
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211.
- AlHarbi, M.; Ali Mobark, N.; AlMubarak, L.; Aljelaify, R.; AlSaeed, M.; Almutairi, A.; Alqubaishi, F.; Hussain, M.E.; Balbaid, A.A.O.; Said Marie, A.; et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist 2018, 23, 1401–1406.
- Yang, C.; Austin, F.; Richard, H.; Idowu, M.; Williamson, V.; Sabato, F.; Ferreira-Gonzalez, A.; Turner, S.A. Lynch syndrome-associated ultra-hypermutated pediatric glioblastoma mimicking a constitutional mismatch repair deficiency syndrome. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003863.
- Shlien, A.; Campbell, B.B.; De Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262.
- Kloor, M.; von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133.
- Bakarurraini, N.A.A.R.; Mutalib, N.S.A.; Jamal, R.; Abu, N. The landscape of tumor-specific antigens in colorectal cancer. Vaccines 2020, 8, 371.
- Saeterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260.
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997.
- Saeterdal, I.; Gjertsen, M.K.; Straten, P.; Eriksen, J.A.; Gaudernack, G. A TGF betaRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother. 2001, 50, 469–476.
- Ripberger, E.; Linnebacher, M.; Schwitalle, Y.; Gebert, J.; Von Knebel Doeberitz, M. Identification of an HLA-A0201-restricted CTL epitope generated by a tumor-specific frameshift mutation in a coding microsatellite of the OGT gene. J. Clin. Immunol. 2003, 23, 415–423.
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors with Microsatellite Instability. Cancer Res. 2020, 80, 3972–3982.
- Westdorp, H.; Gorris, M.A.J.; Boudewijns, S.; Bisseling, T.; de Goede, A.L.; van Rossum, M.M.; Ligtenberg, M.J.L.; Schreibelt, G.; Nagtegaal, I.D.; Figdor, C.G.; et al. Preventive dendritic cell vaccination in healthy Lynch syndrome mutation carriers. Ann. Oncol. 2016, 27, vi362.
- Kloor, M.; Reuschenbach, M.; Karbach, J.; Rafiyan, M.; Al-Batran, S.-E.; Pauligk, C.; Jaeger, E.; von Knebel Doeberitz, M. Vaccination of MSI-H colorectal cancer patients with frameshift peptide antigens: A phase I/IIa clinical trial. J. Clin. Oncol. 2015, 33, 3020.
- Pavelka, Z.; Zitterbart, K.; Nosková, H.; Bajčiová, V.; Slabý, O.; Štěrba, J. Effective Immunotherapy of Glioblastoma in an Adolescent with Constitutional Mismatch Repair-Deficiency Syndrome. Klin. Onkol. 2019, 32.
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338.
- Inderberg, E.M.; Wälchli, S.; Myhre, M.R.; Trachsel, S.; Almåsbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 2017, 6, e1302631.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
23 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No