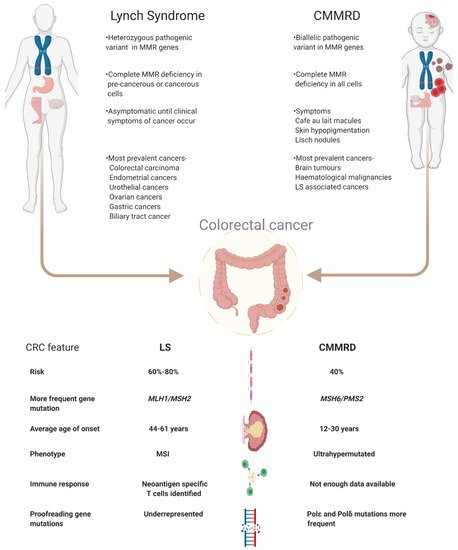
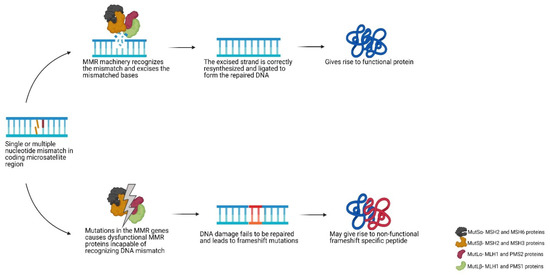
Lynch syndrome (LS) and constitutional mismatch repair deficiency (CMMRD) are hereditary disorders which significantly increase a person’s risk of developing a variety of cancers such as colorectal, endometrial, brain and, for CMMRD also, haematological malignancies. This increased cancer risk is due to inherited mutations in specific types of DNA repair genes, which hampers repair of mispaired or damaged bases during DNA replication. As a consequence, somatic mutations rapidly accumulate and typically include insertions and deletions (indels) in microsatellites that potentially can give rise to neoantigens. These neoantigens open up avenues for neoantigen-targeting immune therapies.
- Lynch Syndrome
- hereditary cancer
- CMMRD
- neoantigen
- colorectal cancer
- mismatch repair deficiency
- targeted therapy
1. Introduction
2. Mismatch Repair Deficiency and Microsatellite Instability
3. Clinical Management of LS and CMMRD
4. Targeting Neoantigens in LS and CMMRD
This entry is adapted from the peer-reviewed paper 10.3390/cancers13102345
References
- Hampel, H.; Hoffner, B. Lynch Syndrome and Immunotherapy. J. Adv. Pract. Oncol. 2017, 8, 7–21.
- Lynch, H.T.; Lanspa, S.; Shaw, T.; Casey, M.J.; Rendell, M.; Stacey, M.; Townley, T.; Snyder, C.; Hitchins, M.; Bailey-Wilson, J. Phenotypic and genotypic heterogeneity of Lynch syndrome: A complex diagnostic challenge. Fam. Cancer 2018, 17, 403–414.
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162.
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793.
- Ricciardone, M.D.; Tayfun, O. Advances in Brief Human MLH1 Deficiency Predisposes to Hematological Malignancy and. Cancer Res. 1999, 59, 290–293.
- Wang, Q.; Lasset, C.; Desseigne, F.; Saurin, J.-C.; Maugard, C.; Navarro, C.; Ruano, E.; Descos, L.; Trillet-Lenoir, V.; Bosset, J.-F.; et al. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum. Genet. 1999, 105, 79–85.
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.A.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.-M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J. Med. Genet. 2014, 51, 355–365.
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565.
- Bucksch, K.; Zachariae, S.; Aretz, S.; Büttner, R.; Holinski-Feder, E.; Holzapfel, S.; Hüneburg, R.; Kloor, M.; Von Knebel Doeberitz, M.; Morak, M.; et al. Cancer risks in Lynch syndrome, Lynch-like syndrome, and familial colorectal cancer type X: A prospective cohort study. BMC Cancer 2020, 20, 1–11.
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise management of ovarian cancer: On the cutting edge. J. Pers. Med. 2020, 10, 41.
- Van Der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.L.; Witjes, J.A.; Hulsbergen-Van De Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; Van Krieken, J.H.J.M.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med. Genet. 2010, 47, 464–470.
- Mankaney, G.; Macaron, C.; Burke, C.A. Refining Risk Factors for Gastric Cancer in Patients With Lynch Syndrome to Optimize Surveillance Esophagogastroduodenoscopy. Clin. Gastroenterol. Hepatol. 2020, 18, 780–782.
- Abedalthagafi, M. Constitutional mismatch repair-deficiency: Current problems and emerging therapeutic strategies. Oncotarget 2018, 9, 35458–35469.
- Lavoine, N.; Colas, C.; Muleris, M.; Bodo, S.; Duval, A.; Entz-Werle, N.; Coulet, F.; Cabaret, O.; Andreiuolo, F.; Charpy, C.; et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J. Med. Genet. 2015, 52, 770–778.
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194.
- Westdorp, H.; Kolders, S.; Hoogerbrugge, N.; de Vries, I.J.M.; Jongmans, M.C.J.; Schreibelt, G. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Lett. 2017, 403, 159–164.
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 1–12.
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 1–14.
- George, J.T.; Kessler, D.A.; Levine, H. Effects of thymic selection on T cell recognition of foreign and tumor antigenic peptides. Proc. Natl. Acad. Sci. USA 2017, 114, E7875–E7881.
- Zhou, J.; Dudley, M.E.; Rosenberg, S.A.; Robbins, P.F. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother. 2005, 28, 53–62.
- Burgess, J.T.; Rose, M.; Boucher, D.; Plowman, J.; Molloy, C.; Fisher, M.; O’Leary, C.; Richard, D.J.; O’Byrne, K.J.; Bolderson, E. The Therapeutic Potential of DNA Damage Repair Pathways and Genomic Stability in Lung Cancer. Front. Oncol. 2020, 10, 1–14.
- Boussios, S.; Moschetta, M.; Karihtala, P.; Samartzis, E.P.; Sheriff, M.; Pappas-Gogos, G.; Ozturk, M.A.; Uccello, M.; Karathanasi, A.; Tringos, M.; et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann. Transl. Med. 2020, 8, 1706.
- Colle, R.; Cohen, R.; Cochereau, D.; Duval, A.; Lascols, O.; Lopez-Trabada, D.; Afchain, P.; Trouilloud, I.; Parc, Y.; Lefevre, J.H.; et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull. Cancer 2017, 104, 42–51.
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179.
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res. 2004, 64, 3014–3021.
- Vos, J.R.; Fakkert, I.E.; Spruijt, L.; Willems, R.W.; Langenveld, S.; Mensenkamp, A.R.; Leter, E.M.; Nagtegaal, I.D.; Ligtenberg, M.J.L.; Hoogerbrugge, N. Evaluation of yield and experiences of age-related molecular investigation for heritable and nonheritable causes of mismatch repair deficient colorectal cancer to identify Lynch syndrome. Int. J. Cancer 2020, 147, 2150–2158.
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346.
- Elze, L.; Mensenkamp, A.R.; Nagtegaal, I.D.; van Zelst-Stams, W.; de Voer, R.M.; Ligtenberg, M.J.L. Somatic Nonepigenetic Mismatch Repair Gene Aberrations Underly Most Mismatch Repair-Deficient Lynch-Like Tumors. Gastroenterology 2021, 160, 1414–1416.e3.
- van Leerdam, M.E.; Roos, V.H.; van Hooft, J.E.; Balaguer, F.; Dekker, E.; Kaminski, M.F.; Latchford, A.; Neumann, H.; Ricciardiello, L.; Rupinska, M.; et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019, 51, 1082–1093.
- Umar, A. Faculty Opinions recommendation of Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Fac. Opin. 2011.
- Leenders, E.K.S.M.; Westdorp, H.; Brüggemann, R.J.; Loeffen, J.; Kratz, C.; Burn, J.; Hoogerbrugge, N.; Jongmans, M.C.J. Cancer prevention by aspirin in children with Constitutional Mismatch Repair Deficiency (CMMRD). Eur. J. Hum. Genet. 2018, 26, 1417–1423.
- Jo, W.-S.; Carethers, J.M. Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomark. 2006, 2, 51–60.
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3219–3226.
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch repair and DNA damage signalling. DNA Repair 2004, 3, 1091–1101.
- Sawant, A.; Kothandapani, A.; Zhitkovich, A.; Sobol, R.W.; Patrick, S.M. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 2015, 35, 126–136.
- Seth, S.; Ager, A.; Arends, M.J.; Frayling, I.M. Lynch syndrome—Cancer pathways, heterogeneity and immune escape. J. Pathol. 2018, 246, 129–133.
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422.
- Bohaumilitzky, L.; von Knebel Doeberitz, M.; Kloor, M.; Ahadova, A. Implications of Hereditary Origin on the Immune Phenotype of Mismatch Repair-Deficient Cancers: Systematic Literature Review. J. Clin. Med. 2020, 9, 1741.
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530.
- Diaz, L.A., Jr.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979.
- Gurjao, C.; Liu, D.; Hofree, M.; AlDubayan, S.H.; Wakiro, I.; Su, M.-J.; Felt, K.; Gjini, E.; Brais, L.K.; Rotem, A.; et al. Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair-Deficient Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 1230–1236.
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779.
- Salman, P.; Panay, S.; Fernández, R.; Mahave, M.; Soza-Ried, C. Evidence of response to pembrolizumab in a patient with lynch syndrome-related metastatic colon cancer. Onco Targets Ther. 2018, 11, 7295–7300.
- Patil, N.R.; Khan, G.N. Exceptional response to a single cycle of immunotherapy in a lynch syndrome patient with metastatic pancreatic adenocarcinoma. Am. J. Case Rep. 2020, 21, 1–6.
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211.
- AlHarbi, M.; Ali Mobark, N.; AlMubarak, L.; Aljelaify, R.; AlSaeed, M.; Almutairi, A.; Alqubaishi, F.; Hussain, M.E.; Balbaid, A.A.O.; Said Marie, A.; et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist 2018, 23, 1401–1406.
- Yang, C.; Austin, F.; Richard, H.; Idowu, M.; Williamson, V.; Sabato, F.; Ferreira-Gonzalez, A.; Turner, S.A. Lynch syndrome-associated ultra-hypermutated pediatric glioblastoma mimicking a constitutional mismatch repair deficiency syndrome. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003863.
- Shlien, A.; Campbell, B.B.; De Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262.
- Kloor, M.; von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133.
- Bakarurraini, N.A.A.R.; Mutalib, N.S.A.; Jamal, R.; Abu, N. The landscape of tumor-specific antigens in colorectal cancer. Vaccines 2020, 8, 371.
- Saeterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260.
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997.
- Saeterdal, I.; Gjertsen, M.K.; Straten, P.; Eriksen, J.A.; Gaudernack, G. A TGF betaRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother. 2001, 50, 469–476.
- Ripberger, E.; Linnebacher, M.; Schwitalle, Y.; Gebert, J.; Von Knebel Doeberitz, M. Identification of an HLA-A0201-restricted CTL epitope generated by a tumor-specific frameshift mutation in a coding microsatellite of the OGT gene. J. Clin. Immunol. 2003, 23, 415–423.
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors with Microsatellite Instability. Cancer Res. 2020, 80, 3972–3982.
- Westdorp, H.; Gorris, M.A.J.; Boudewijns, S.; Bisseling, T.; de Goede, A.L.; van Rossum, M.M.; Ligtenberg, M.J.L.; Schreibelt, G.; Nagtegaal, I.D.; Figdor, C.G.; et al. Preventive dendritic cell vaccination in healthy Lynch syndrome mutation carriers. Ann. Oncol. 2016, 27, vi362.
- Kloor, M.; Reuschenbach, M.; Karbach, J.; Rafiyan, M.; Al-Batran, S.-E.; Pauligk, C.; Jaeger, E.; von Knebel Doeberitz, M. Vaccination of MSI-H colorectal cancer patients with frameshift peptide antigens: A phase I/IIa clinical trial. J. Clin. Oncol. 2015, 33, 3020.
- Pavelka, Z.; Zitterbart, K.; Nosková, H.; Bajčiová, V.; Slabý, O.; Štěrba, J. Effective Immunotherapy of Glioblastoma in an Adolescent with Constitutional Mismatch Repair-Deficiency Syndrome. Klin. Onkol. 2019, 32.
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338.
- Inderberg, E.M.; Wälchli, S.; Myhre, M.R.; Trachsel, S.; Almåsbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 2017, 6, e1302631.