 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karthik Shankar | + 6888 word(s) | 6888 | 2021-05-21 08:43:51 | | | |
| 2 | Rita Xu | -1 word(s) | 6887 | 2021-05-24 05:13:38 | | |
Video Upload Options
Hot carriers are electron-hole pairs formed by Landau damping or chemical interface damping of particle plasmons. The term “hot electrons” not only describes the individual electrons themselves, but also describes the Fermi–Dirac distribution of electrons in a solid albeit with an elevated effective temperature—the effective temperatures involved when considering the carrier kinetic energies and carrier densities in the solid, and not that of the solid itself- as opposed to thermal equilibrium. Typically, hot carriers lose their excess energy and relax to thermal equilibrium within a few picoseconds of formation due to a variety of scattering processes. Therefore, it is very difficult to extract useful work from hot carriers with thermodynamic efficiencies that are of interest for practical devices. However, heterojunctions of TiO2 with coinage metals have been consistently shown to be effective in utilizing hot carriers to perform useful work, such as driving a chemical reaction or generating an electric current. A secondary benefit of such TiO2-coinage metal heterojunctions is the "visible light sensitization" effect which allows broader harvesting of visible light.
1. Introduction
Solar energy is among the cleanest, and most abundant renewable energy sources available to the world. Our planet exploits solar energy routinely through photosynthesis—the process by which plants, algae, photosynthetic bacteria, and protists capture sunlight, water, and residual CO2 in our atmosphere as reactants for water-splitting chemistry:
6H2O + 6CO2 + Sunlight → C6H12O6 + 6O2
This process allows for the decomposition of water to molecular oxygen, and the transformation of CO2 to carbohydrates and other carbon-rich products integral to the sustainability of our planet’s biosphere [1].
Amidst the rapidly rising global energy demand (17.4 Terrawatts (TW) in 2015 and a 2.2% growth averaged in 2017, the fastest since 2013) [2] and environmental crises, the efficient utilization of solar energy in chemical transformations is extremely important for the modern energy industry. Global energy consumption is predicted to increase to about twice the current value in 2050 [3][4]. For decades, fossil fuels such as oil, coal, peat, and natural gas have served as conventional energy sources to meet the world’s energy demands and have provided for sustainable economic development. 140,000 TWh of energy per year is consumed by mankind with more than 80% accounted by fossil fuels. The dominance of fossil fuels in the global energy generation and distribution infrastructure is largely due to their availability, stability, and high energy density [5] but the proliferation of fossil fuel burning has led to a dramatic increase in atmospheric CO2 levels over the last century (up to 100 parts per million by volume) [6] with CO2 emissions widely considered as the major cause of global warming. Notwithstanding the increasing need to mitigate this global crisis, fossil fuels are also a limited energy source.
To address these issues, considerable effort has been placed on the development of renewable, environment-friendly, artificial photosynthetic technologies to sustain modern technological civilization. The use of artificial photosynthetic technology is a means to not merely mimic photosynthesis but to improve our knowledge of the process and enhance it to our selective needs through artificial means (Figure 1). Solar irradiation on our planet in just one hour exceeds our annual energy consumption. By tapping into even 0.02% of the incoming solar energy, we could satisfy all our current energy needs [7][8]. In this objective, an assortment of technologies has been developed ranging from biological systems (algae), inorganic photocatalysts (transition metal oxides or semiconductors, particularly TiO2-based catalysts), organic photocatalysts (metal-organic complexes), biomimetic systems (enzyme-activated or dye-sensitized semiconductors), tandem cells, and z-schemes to name a few [6][9].
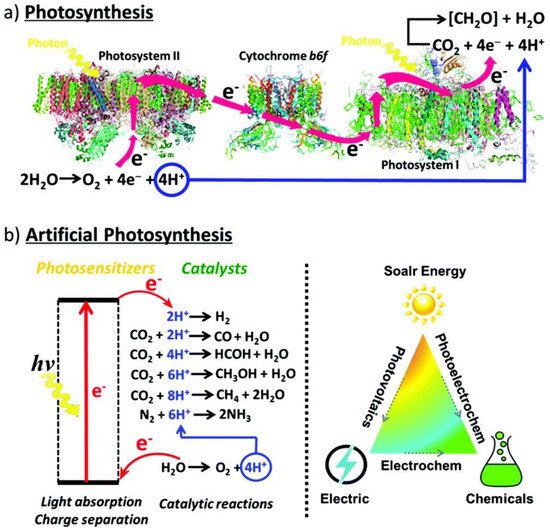
Figure 1. (a) Photosynthesis is enabled through the collaborative efforts of two photosynthetic complexes, PSI and PSII, where PSI serves as the reaction center and light harvesting complex and PSII is the site of water oxidation. Thus, H2O is oxidized in PSII into O2 releasing four protons and electrons, respectively, that are transferred via cytochrome b6f, an enzyme in plant chloroplasts, to PSI where they are consumed by CO2 reduction to produce carbohydrates. (b) Artificial photosynthetic systems for photocatalysis are being developed to mimic and provide for the very same conversion of solar energy through alternative energetic pathways and selectivity for fundamental and desirable chemical reactions, including water splitting, CO2 photoreduction, and the degradation of harmful organic pollutants. Reprinted with permission from Ref [10] with attribution and adherence to Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Copyright Royal Society of Chemistry (2019).
Over the recent decade, semiconductor photocatalysts have become highly popular as the key artificial photosynthetic technology and have set the basis for research in the field of photocatalysis. Researchers have developed many semiconductors as suitable candidates for photocatalysts including metal oxides, metal chalcogenides, metal nitrides, bismuth oxyhalides, carbon nitrides, and III-V compounds [11][12][13][14][15][16][17][18][19][20][21][22][23][24][25]. Semiconductor photocatalysts absorb photons to generate active electrons and holes that are then utilized to initiate chemical reduction and oxidation reactions [26][27][28]. A viable photocatalyst, in general, must allow for optimal light absorption (wide light-absorption range), efficient charge separation (good band energetics), charge migration for necessary chemical reactions (high carrier mobility and long carrier diffusion lengths), as well as strong catalytic activity, stability, and from a commercial viewpoint, high sustainability and low cost.
TiO2 has been the poster-child for semiconductor photocatalyst materials due to its relatively low cost, high availability, low toxicity, stability in both acidic and basic media, and resistance to photo-corrosion [29]. Yet, large-scale commercialization of semiconductor photocatalytic technology in the environmental and energy industries is still at its advent and remains to be fully exploited. This is because despite their obvious advantages, critical and debilitating material-sensitive limitations have surfaced over the years concerning semiconductor photocatalysts. Popular photocatalysts, such as TiO2 and SrTiO3, are relatively cheap, easy to process, and durable but are poor absorbers of visible light due to their wide bandgaps [30]. Contrastingly, narrow bandgap semiconductors, such as Si and Cu2O, lack long-term catalytic efficiency, and photo-corrode easily. Other semiconductors, such as Fe2O3, are inhibited by extremely low photocatalytic activity [30].
To overcome these limitations, plasmonic photocatalysts have emerged as a promising technology for harvesting and converting solar energy [31][32][33][34][35]. This is achieved by the generation and transfer of energetic charge carriers or “hot electrons” via resonant interaction of incident light with the collective and coherent motion of electrons in metal nanostructures to initiate, enhance, and promote photocatalytic activity. The exploitation of hot electrons produced by the localized surface plasmon resonance (LSPR) of noble metal nanoparticles in photocatalysis and photovoltaics has recently witnessed a surge of research interest [36][37][38][39][40][41][42]. The research interest is well-deserved since optimal exploitation of hot electrons holds out the promise of high performance, durable photocatalysts for water treatment, solar hydrogen generation from water splitting, and CO2 photoreduction. In spite of such intense research interest, many aspects related to the fundamental physics of hot electron generation and transfer from particle plasmons remain unclear. Our review is comprehensive and incorporates information from a broad cross-section of recent articles. Since the authors of this work have been researching the topic of noble metal nanoparticle, TiO2 heterojunction photocatalysts for water-splitting and CO2 photoreduction, we are well-placed to discuss the latest developments in this fast-changing field. One unique aspect of our review is that it has a self-contained section on “Probing Hot Electrons”, where we discuss in great detail the application of different spectroscopic techniques to characterize plasmonic hot electron photocatalysts and the interpretation of the characterization data thus obtained. We strongly believe this information will be valuable to both new researchers entering the field and even to experienced researchers who might have not considered a technique outside the suite of techniques they’re comfortable with.
1.1. What Are Hot Electrons and Why Are They Important?
Theoretical work on hot electrons began in the 1930s. Hot electrons can be generated by applying a strong electric field to a conductor. For metals, high electric fields may cause melting or result in extremely high joule heating. Therefore, most early hot electron work focused on understanding dielectric breakdown in insulators [43].
Hot electrons are essentially electrons that are not in thermal equilibrium with their immediate environment (generally, the atoms comprising a material) [43]. These electrons have a very high effective temperature (as high as several thousand Kelvin) compared to room temperature, due to their kinetic energy or resonant interaction/coupling with light. Hot electron lifetimes vary with respect to the relevant material structures. Hot electrons in bulk gold with an energy greater than 1 eV above the Fermi Energy (EF) have a lifetime smaller than 50 fs with the dominant relaxation mechanism being inelastic electron–electron interactions [44]. Meanwhile, due to reduced electron–electron interactions and confinement effects in small gold nanoparticles (Au NPs), hot electron lifetimes in Au NPs are typically an order of magnitude larger, in the range of 100–500 fs [45].
Both hot electrons and their counterparts in hot holes can be very effective in stimulating chemical and physical processes, being only limited by the rapid relaxation processes that accompany their emission. This very relaxation of the high energy carriers also helps stimulate heating of the solid structures involved. This fits the paradigm of photocatalysis as hot electrons can be utilized for various effects from local heating of particles and reactants to photochemistry, photodesorption, and controlled chemical reactions [46][47][48][49]. The discoveries of photochemical water splitting on TiO2 electrodes using ultraviolet light [50], surface-enhanced Raman spectroscopy (SERS) [51], and femtochemistry studies on single-crystal metal surfaces [52][53][54][55] served as foundational steps towards current interest in the utilization of hot electron induced chemical reactions on photoexcited metal surfaces, more precisely identified as plasmonic hot electron photocatalysis.
1.2. Plasmonic Hot Electron Photocatalysis Using TiO2–Noble Metal Nanostructures
Hot electron photocatalysts are typically composite systems that incorporate a semiconductor with a plasmonic noble metal nanostructure in a heterojunction. Plasmonic noble metal nanostructures have electron densities that can couple with wavelengths of electromagnetic radiation (in the visible spectral range) that are far larger than the nanostructure itself due to the dielectric-metal interface between the particles and the surrounding medium; contrastingly, in pure metals, there is a maximum limit on the magnitude of wavelengths (work-function dependent) that can effectively couple with the material sizes involved [56]. Plasmonic noble metal nanospheres have commonly been utilized as hot electron photocatalysts although recently diverse nanostructures, such as nanocubes, nanorods, nanoshells, gap plasmon structures, etc., have also been investigated [57][58][59][60].
Plasmon-enhanced photocatalysis capitalizes on the resonant interaction of light with the collective and coherent motion of electrons in the noble metal nanostructure allowing for their ability to focus light into small volumes and thus generate large enhancements in the amplitude of the local electromagnetic field [61]. This resonant interaction is also used to perform chemical reactions. Hot electrons, in the context of photocatalysis, can holistically be defined as electrons with energies >> kT above the Fermi level on optically excited plasmonic nanoparticle surfaces that are then transferred to a medium (be it a chemical adsorbate, a semiconductor, or even the surrounding environment) where they perform a particular function (in photocatalysis, a chemical reaction). In this manner, plasmonic photocatalysis allows for the manipulation of light with nanometer-scale precision, and for reaction control of hot carrier processes at sub-femtosecond timescales [46]. Nearly all semiconductor-based hot electron photocatalysts demonstrated until now consist of Schottky junctions of n-type semiconductors with plasmonic noble metals (Figure 2). Figure 2 indicates that on the metal side, a fraction of electrons with energies exceeding the Schottky barrier height are able to cross over into the semiconductor side of the junction. Figure 2 shows that photogenerated holes in the semiconductor drift towards the metal because of the built-in field associated with the Schottky junction. There is a negligibly small equilibrium concentration of holes on the semiconductor side of the junction due to which the hot electrons that do cross over from the metal would be expected to have long lifetimes in the semiconductor, due to the lack of recombination events. In lifetime semiconductors, such as Si, Ge, InP, etc., charge neutrality will be restored in a duration roughly comparable to the dielectric relaxation time (<100 ns) [62]. In relaxation semiconductors, such as TiO2, ZnO, GaN, SrTiO3, etc., the dielectric relaxation times are orders of magnitude larger than in lifetime semiconductors due to which injected hot electrons can have unusually long lifetimes of milliseconds to even hours, which allows abundant time for these electrons to reduce reactant species [42][62][63][64]. Au/n–TiO2 plasmonic noble metal nanostructure–semiconductor heterojunctions are particularly ubiquitous. There is emerging interest in using p-type TiO2/noble metal heterojunctions to achieve enhanced photocatalytic performance by enabling the fast injection (and subsequent utilization) of hot holes into TiO2 before appreciable thermalization. The basic motivation for the use of such heterojunctions lies in the asymmetric energy distribution of hot carrier pairs produced through Landau damping of the particle plasmon. In Au, the hot carriers produced consist of high energy holes and low energy electrons, i.e., the hot holes are much hotter than the hot electrons, making it more worthwhile to drive chemical reactions using hot holes [65][66]. Anodization and sol-gel synthetic strategies that combine a high density of Ti3+ defect states together with elevated temperature oxygen annealing to reduce oxygen vacancies are known to produce p-type TiO2 [67][68]. Jinhua Ye and colleagues constructed a Schottky junction consisting of p-type TiO2 decorated with Au NPs and observed a remarkable fivefold enhancement of the acetone evolution rate in the photocatalytic degradation of isopropanol. Likewise, Y. Zhang et al. [69] observed a significant enhancement in the photocatalytic degradation of tetrabromobisphenol A using Ag-loaded p-type TiO2.
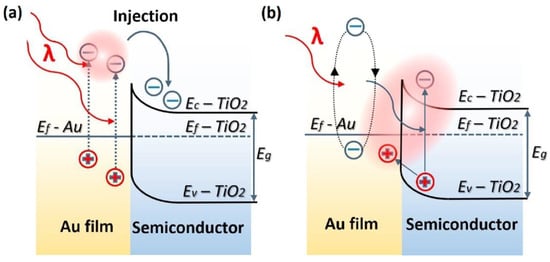
Figure 2. Energy band-diagram of Au and n-type TiO2 heterojunction showing LSPR-driven hot electron injection from Au into TiO2 by (a) over barrier thermionic emission and (b) tunneling mechanism. Note the bending of the conduction and valence bands of TiO2 at the contact interface of the two materials due to the equilibration of Fermi levels upon contact forming a Schottky barrier. EF, EVB, ECB, ϕB, and L are the Fermi level, valence band level, conduction band level, Schottky barrier height, and the width of depletion layer, respectively. Reprinted with permission from Ref [70] Copyright Elsevier (2017).
As mentioned earlier, when it comes to semiconductor photocatalysts, TiO2 remains the benchmark thanks to its relatively low cost, high availability, low toxicity, stability in both acidic and basic media, and resistance to photo-corrosion [29]. TiO2 has a wide band gap (3.2 eV for anatase and 3.0 eV for the rutile phase) and a relatively high absorption coefficient for ultraviolet photons [71]. TiO2 has a minority carrier (hole) diffusion length of 70 nm for anatase TiO2 and 10 nm for the rutile phase [72][73]. Due to its large bandgap, TiO2 primarily absorbs UV photons and taps into merely 5% of the solar energy that our planet receives. Although doping can extend the light absorption range of TiO2 from the UV to visible wavelengths, the absorption coefficient and the photocatalytic activity of TiO2 typically decrease [9][29][73][74]. To compensate for these limitations, various architectures of TiO2 photocatalysts ranging from powders in aqueous solutions [74], nanoparticles (0D), nanorods and nanotubes (1D), nanosheets and films (2D), and 0D-1D-2D integrated nanostructures (3D) (Figure 3) have been investigated [9]. 0D structures have the highest surface area per unit mass, a beneficial feature for catalysis, but have the disadvantages of not being able to sustain an internal electric field and confining both electrons and holes in a small volume of space until charge separation occurs. 1D structures combine a high surface area and the possibility of intra-nanowire charge separation due to a built-in field with the orthogonalization of light absorption/charge generation and charge separation processes [75][76]. Other efforts have focused on varying crystalline phase systems (rutile, anatase, and brookite) [77][78][79], doped heterojunctions, and mesoporous supports [80][81] with the focus being to optimize integral nanoscale properties, such as the optical path length, carrier mobility [82][83], charge carrier kinetics [84], light absorption [85], band bending, etc. Despite all of this, definitive success in sensitizing the photocatalytic activity of TiO2 to visible wavelengths is yet to be achieved.
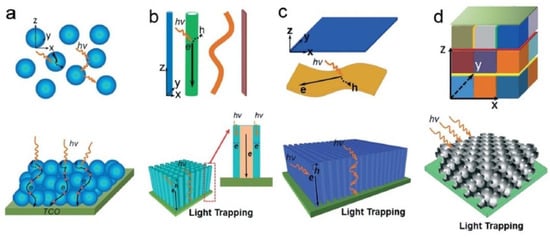
Figure 3. Various nanoscale architectures that can be used in photocatalytic applications from (a) 0D nanocrystals, (b) 1D nanostructures, (c) 2D nanosheets and films, and (d) 0D-1D-2D integrated 3D nanostructures. The figure also illustrates the light scattering, light trapping, and charge transport processes in the corresponding nanostructures. Reprinted with permission from Ref [9] Copyright 2015 Royal Society of Chemistry. Figure 3d originally adapted by Ref [9] from Ref [86] with permission from John Wiley, and reprinted here with permission from John Wiley. Copyright John Wiley and Sons (2013).
This is where plasmonic photocatalytic systems incorporating the use of TiO2–plasmonic noble metal heterojunction nanostructures enter as a viable and realistic solution for extending the photoresponse of TiO2; hot electrons are at the core of this development. The family of plasmonic noble metals is small with gold (Au) and silver (Ag) being the two most recognized elements. Using the knowledge that Au and Ag nanostructures both have low loss surface plasmon resonances excited by visible and near-infrared photons, one can promote and mediate the charge transfer of hot electrons to the neighboring TiO2 semiconductor which can be utilized as a secondary surface or port for photocatalytic reactions in addition to photocatalytic reactions occurring on the surface of the noble metal. In a standard system, when placed in intimate contact, Au and n-type TiO2 form a Schottky junction. Noble metals have a high work function, and in the case of Au, its Fermi level is located below that of n-type TiO2. Upon contact, the Fermi levels equilibrate resulting in the bending of the conduction band of TiO2 and the formation of a Schottky barrier (Figure 2). Thus, a depletion region is formed, where an internal electric field is maintained and directed from TiO2 to Au. Upon excitation by incident visible light, it is this electric field that drives the motion of photogenerated electrons in the depletion region to move to TiO2 and holes to Au, thus preventing recombination. These electrons and holes are the ones to participate in photocatalytic reactions [30].
Plasmon-mediated electron transfer involving the injection of hot electrons across a Schottky barrier from Au NPs into the conduction band of TiO2 is known to occur at timescales of 250 fs or shorter [87][88], and the transferred electrons have been observed to exhibit unusually long lifetimes (>103 s) in rutile TiO2, larger by two orders of magnitude than the lifetimes of photoexcited carriers generated directly in TiO2 [64]. The importance of nanostructured Au NP–TiO2 heterojunctions lies in the fact that hot electrons formed in Au NPs by decay of plasmons stimulated by visible photons with energies well below the bandgap of TiO2, have been demonstrated to drive chemical reactions subsequent to injection across the Schottky barrier into TiO2. Thus, heterojunctions of plasmonic nanoparticles with TiO2 enable visible light sensitization. The sensitization effect can be maximized in geometries where the plasmon-mediated local electromagnetic field enhancement at the metal–semiconductor interface is large, i.e., at hot spots [89][90].
A key question relates to the theoretical maximum power conversion efficiency (PCE) achievable in a plasmonic hot electron cell. White and Catchpole demonstrated that for a typical parabolic density of states (DOS) in the conduction band (CB) of the metal, the PCE was capped at 7.2%, rising however to 22.8% if the CB DOS could be engineered such that electrons close to the Fermi level (EF) were preferentially excited over lower energy electrons during the non-radiative decay of the particle plasmon [91]. This calculation assumed the sequential mechanism of plasmon decay and did not take into account more direct hot carrier formation and separation mechanisms, such as chemical interface damping. It is important to note that there are no lab-scale or commercially deployed photocatalysts that can convert sunlight into chemical fuels over extended durations with PCEs of over 5%. Therefore, while the PCEs potentially achievable using plasmonic hot electron devices might seem unremarkable for photovoltaics where lab-scale PCEs of 20–25% are routinely obtained for single junction silicon, CdTe, and halide perovskite solar cells, the same PCEs if achieved in the context of photocatalysis, would constitute a dramatic enhancement over the state of the art.
Au and Ag remain the most popular plasmonic noble metals in use. Ag is an ideal material for plasmonics, due to its low optical loss in the visible and NIR spectral ranges [92]. Au performs equivalently well in the visible and NIR spectral ranges and is also chemically superior to Ag which oxidizes under ambient conditions. Various other plasmonic noble metals have been considered in the field including Cu, Al, Pt, Pd, etc. Surface plasmons form at visible and near-infrared wavelengths in the base metals Al and Cu. However, the much larger dielectric losses (due to both radiation- and interband damping in Al [93] and interband damping in Cu [94]) result in broad, low quality factor resonances with a weak local field enhancement and insufficient production of usable hot carriers. Furthermore, Cu and Al are chemically unstable under atmospheric conditions. These reasons limit the use of Cu and Al to niche applications that exploit the LSPR resonances of Cu and Al in the IR and UV spectral ranges. Pd and Pt exhibit very strong interband damping [93], and have attracted attention in plasmonic catalysis, largely due to their catalytic abilities, and are often incorporated in bimetallic plasmon systems, due to their weak absorption at visible wavelengths. Beyond this, various studies have been conducted over the years to extend and diversify the library of plasmonic materials that could be utilized for plasmonic applications [95]. The alloying of different noble metals has been an alternative to tune the LSPR wavelength [96][97]. Similar approaches have also been considered in the fabrication of bimetallic and trimetallic systems where plasmonic noble metals are fabricated in conjunction with a catalytic metal with the former serving as a nanoantenna and the other as a catalytic medium [60]. Efforts have also been made to modulate the LSPR behavior of noble metals via exotic morphologies [98]. Atomistic and continuum calculations have provided deeper understanding of the plasmonic responses of these noble metals, and recent efforts have also focused on the use of phase and compositional changes to help evoke plasmon responses in lower cost, non-plasmonic noble metals, transition metal oxides and nitrides, and chalcogenide compounds [21][99][100][101][102][103].
The extraction of hot electrons from plasmonic nanoparticles would be useful in a variety of applications, including cancer tissue targeting [104][105][106], lasing [107][108], imaging [109][110], molecular characterization [51][111][112], and solar energy conversion (solar cells and photovoltaics) [113][114][115][116]. Some of these applications focus on the design of plasmonic nanostructures that optimize the confinement, bending, and propagation of light while minimizing internal losses, while others focus on charge carrier formation and transfer processes [117]. Plasmonic photocatalysis applications fall in the latter category. In fact, Au nanoparticles coupled to TiO2 for water splitting constitute the earliest examples of plasmon-enhanced photocatalytic and energy conversion systems. It is critical to understand the inherent nature and origin of these microscopic high energy charge carriers for their efficient implementation in photocatalytic systems. Gaps in knowledge and understanding persist within the scientific community regarding the origin of hot electrons in optically excited plasmonic nanoparticles, and the subsequent charge transfer dynamics that take place within said systems to promote photocatalytic activity.
2. Digging Deeper into Hot Electrons
Early seminal research on semiconductor devices, and the physical modeling of extended metal surfaces set the foundation for our current understanding of hot electron phenomena. The term “hot electrons” not only describes the individual electrons themselves, but also describes the Fermi–Dirac distribution of electrons in a solid albeit with an elevated effective temperature—the effective temperatures involved when considering the carrier kinetic energies and carrier densities in the solid, and not that of the solid itself- as opposed to thermal equilibrium [118].
2.1. Surface Plasmons
Hot electrons are now a major focus in the field of plasmonics, which is the study of the interaction of light with free electrons in a metal. Hot electrons are generated via surface plasmon (or plasmon) excitation. Surface plasmons are the quantized collective and coherent oscillations of free electrons in a metal in response to excitation by incident photons at a metal-dielectric interface [119]. The electric field of the incident light guides the collective oscillations of these free electrons resulting in two characteristic modes: Surface Plasmon Polaritons (SPPs) and Localized Surface Plasmon Resonances (LSPRs). These resultant modes are largely determined by the morphology of the metallic structures that enable them (Figure 4) [30].
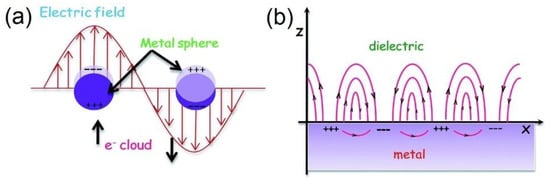
Figure 4. Schematic illustrations of (a) Localized Surface Plasmon Resonances and (b) Surface Plasmon Polaritons. Note the differences in morphologies of the structures involved. LSPRs are excited on metal nanostructures smaller than the electron mean free path within the material as well as smaller than the wavelength of incident light, such as the nanospheres in (a) where free electrons are displaced from the positive ions, driven by the propagating electric field component of the incident light, and oscillate collectively in resonance. In (b) the metal surface’s characteristic dimension is larger than the wavelength of incident light resulting in the excitation of a propagating surface plasmon polariton that travels along the surface with evanescent waves that diminish perpendicular to the surface. Reprinted with permission from Ref [120] Copyright Royal Society of Chemistry (2016).
The excitation of SPPs occurs predominantly in continuous metal structures with characteristic dimensions larger than the incident wavelength of light. The corresponding plasmon oscillations propagate primarily along the metal surface for distances of tens to hundreds of micrometers, while declining as evanescent waves perpendicular and away from the metal surface [121]. In the opposite extreme, for metal nanostructures that are smaller than the electron mean free path within the material and the incident wavelength of light, LSPRs are generated [30]. Here, the collective oscillation of the free electrons at the metal-dielectric interface is driven by the electric field of the incident light with a resonance being achieved when the frequency of the incident radiation matches the oscillation frequency of the free electrons in the metal nanostructure. While SPPs are representative of propagating or traveling plasmons, LSPRs are characterized by non-propagating or standing wave plasmons confined strictly within the boundaries of the metal nanostructure. LSPRs can be excited on metal nanoparticles of various geometries including spheres, prisms, cubes, shells, etc., as well as around nanoholes or nanorods or nanoscale corrugations in thin metal films [122].
The resonant interaction observed in LSPRs is the main factor towards the confinement of photonic energy to the surface of the nanostructure for a duration that exceeds the time-scales photons would spend in the same volume traveling at the speed of light [123]. Consequently, there is an amplification of the local electric field of the incident light as well as the formation of a high concentration of energetic electrons at the surface of the nanostructure. In conclusion, plasmons can be understood as charge density oscillations (non-propagating LSPRs or propagating SPPs) that are a result of dipole and higher order multipole formation in the metal structures described above, due to incident electromagnetic wave excitation.
2.2. Sequential Mechanism of Hot Electron Relaxation
Hot electrons proliferate various critical technologies that take advantage of the LSPR mechanism. In the context of plasmonic photocatalysis, the focus is largely on LSPRs in plasmonic nanoparticles rather than SPPs, unless mentioned otherwise. LSPR excitation can be used to drive remote and direct photochemistry; photonic energy can either be transferred to nearby semiconductors, metals, and molecular photocatalysts or facilitate chemical transformations that occur directly on the surface of the plasmonic nanostructures [123]. All these processes are characterized exclusively by electron or hole transfer from excited metal nanoparticles to acceptor states in semiconductors or molecules. In explaining the charge transfer dynamics involved, the scientific community has, for a large part, been guided by the well-established work on the physical modeling of extended metal surfaces.
Conventional theory suggests a sequential charge excitation/transfer process (Figure 5a) to explain charge transfer dynamics when LSPR excitation occurs on a plasmonic metal nanostructure. Here, a photon excites the LSPR of a nanoparticle to form plasmons, which dephase nearly instantaneously to yield excited electron-hole pairs. The non-radiative decay of plasmons into electron-hole pairs can involve either intraband transitions or interband transitions [124]. A competing mechanism is the radiative decay of particle plasmons. Dephasing refers to the reduction in the amplitude of coherent motion of electrons, which in turn depends on the strength of the coupling of the plasma oscillation to the electron-hole continuum [125]. Dephasing that involves the loss of energy from the collective oscillation of free electrons to the excitation of individual electron-hole pairs is also known as Landau damping [126]. In ~20 nm sized nanoparticles formed by electron beam lithography, single particle near-field scanning optical microscopy (NSOM), and second harmonic generation (SHG) spectroscopy have been used to measure dephasing times of 4–8 fs in Au NPs and 7–10 fs in Ag NPs [125][127]. The electron-hole pairs produced in a few femtoseconds, due to dephasing, are distributed over a range of energies allowing higher energy charge carriers to occupy acceptor states in nearby semiconductors or molecules. The subsequent cooling of the energetic electron-hole pairs to yield a Fermi–Dirac distribution of electrons at an elevated temperature occurs over tens to hundreds of femtoseconds with contributions from electron–electron scattering in the bulk, radiative damping, and electron-surface collision damping (Figure 5a) [61]. Further relaxation of the carrier distribution occurs through collisions with phonons over a few picoseconds, as shown in Figure 5a, while the excited phonons equilibrate over hundreds of picoseconds. A kinetic representation of these damping processes is used in time-resolved studies involving femtosecond pulse experiments where electrons are observed to be excited via low-energy and high-energy transitions. The distribution of electrons during the femtosecond-pulse would include a large population of low-excitation energy electrons and some high-excitation energy electrons. Considering the relaxation time scales involved in the excitation of the LSPR, where electron–electron scattering occurs in ~100 fs [128][129] followed by electron–phonon coupling at the picosecond timescale, the conventional description of LSPR excitation on a plasmonic nanoparticle says that there are significantly few highly excited electrons in the final thermal electron distribution compared to the initial excited distribution.
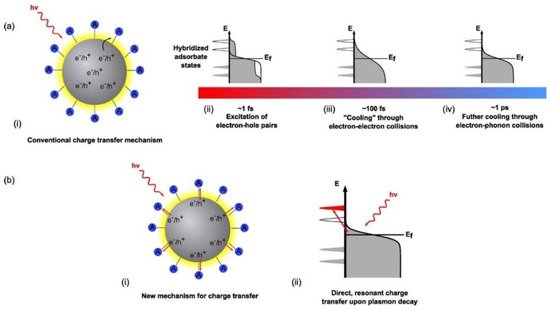
Figure 5. Modes of charge transfer and relaxation mechanisms in metal nanostructures. Ref [117] In the conventional charge transfer mechanism (a), resonant photon absorption creates hot electron-hole pairs within the metal nanostructure. What begins as an equilibrium thermal distribution of charge carriers in the metal nanostructure rapidly changes (i) to a nonequilibrium athermal hot electron distribution (~1 fs) (that cannot be described by Fermi–Dirac statistics) (ii) Hot electrons are now continuously transferred to the conduction band of the semiconductor from the tail portion of the electron distribution of the noble metal (iii) This athermal distribution rapidly dephases or cools through electron–electron collisions taking place on the order of ~100 fs. (iv) Further cooling through electron phonon collisions occurs on the order of ~1 ps resulting in the thermalization of the initial athermal distribution and a subsequent relaxation towards equilibrium. Alternatively, in (b) there is the Dissociation Induced Electron Transfer (DIET) mechanism, where electrons generated under excitation are directly injected into the conduction band of the semiconductor without and before any further interactions with other electrons. (ii) It is a direct, resonant transfer of charge carriers that circumvents the thermalization and relaxation mechanisms of hot electrons depicted in the sequential mechanism in (a). Reprinted with permission from Ref [117] Copyright American Chemical Society (2016).
As a consequence of LSPR excitation, two types of charge carriers can be distinguished: (i) low-excitation energy charge carriers (also identified as Drude electrons and holes) near the Fermi level, and responsible for plasmon oscillations, and (ii) high-excitation energy charge carriers (the “hot electrons”) with energies >> kT above the Fermi level. The prerequisite for plasmonic photocatalysis is the efficient extraction of these hot electrons to help support or drive photocatalytic reactions but, according to the conventional theory stated above, the expected yield of hot electrons is low. This is because a large fraction of the formed energetic charge carriers lacks sufficient energy to support photocatalytic activity or energy transfer reactions, and most of the charge carrier energy is immediately lost upon LSPR dephasing through interactions with other electrons and phonons within the nanoparticle.
2.3. Alternative Mechanisms of Hot Electron Relaxation
In the sequential excitation and relaxation picture, the key to efficient plasmonic photocatalysis lies in extracting hot carriers before they fully equilibrate. However, there have been multiple experimental observations reporting fundamental deviations from the conventional description of the charge excitation/transfer mechanism, most notably concerning semiconductor-to-adsorbate charge transfer reactions. The extraction process involves tunneling through or thermionic emission of the hot carriers over the Schottky barrier into the semiconductor, as shown in Figure 2. Considering the number of hot carriers with sufficient energy and momentum to cross the barrier, the nature of the carrier distribution, the probability that hot carriers will reach the semiconductor-noble metal interface and the transmission probability across the interface, the sequential mechanism dictates that injection efficiencies of ~1% are expected [130]. However, hot electron injection efficiencies of 20–50% for Au NP−TiO2 NP heterojunctions have been observed by multiple research groups using femtosecond transient absorption spectroscopy [130][131]. The sequential excitation-relaxation/transfer picture also requires hot carriers to be extracted from the metal by the semiconductor at timescales shorter than a few hundred femtoseconds. Such ultrafast charge transfer at timescales of 50–240 fs following excitation using 550 nm photons (close to LSPR of gold spheres) has indeed been observed in Au NP−TiO2 NP heterojunctions [132][133].
A completely different mechanism for hot electron harvesting has been suggested involving the direct excitation of interfacial charge transfer (IFCT) states (Figure 6b). The IFCT mechanism refers to plasmon induced metal-to-semiconductor interfacial transitions (PICTT) [134] where it is postulated that the noble metal plasmon as well as the strong coupling and mixing of metal and semiconductor levels allow for the direct generation of an electron in the semiconductor and a hole in the noble metal (Figure 6c). While plasmon-induced IFCT was demonstrated in Au NP−CdSe heterojunctions [134] and has long been implicated in the anomalously high visible response observed in nanostructured Cu–TiO2 heterojunction photocatalysts [135][136][137][138][139][140][141], direct excitation of charge transfer states is more commonly found in experiments with the addition of adsorbates to the surfaces of plasmonic nanostructures. It is well known that molecule-to-semiconductor electron-transfer reactions can occur at sub-picosecond timescales [142]. This is primarily due to the high density of vacant acceptor states in the semiconductor and invocation of Fermi’s golden rule. Contrastingly, the reverse charge-transfer reaction from semiconductor-to-molecule has been shown to be quite slow, due to the much lower density of states in the molecules [143]. Therefore, the possibility for a significant amount of hot electron transfer from the noble metal to vacant molecular states, according to the conventional theory described above, would be highly unlikely.
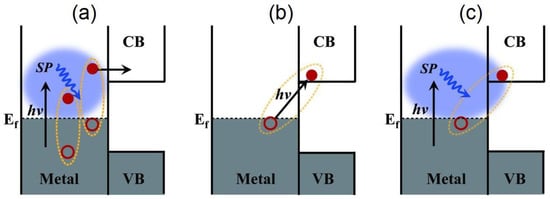
Figure 6. A perspective on the various charge-separation pathways in noble metal–semiconductor systems. (a) The conventional plasmonic hot electron transfer (PHET) mechanism where a plasmon (blue ellipsoidal cloud) in the noble metal dephases into a hot electron-hole pair via Landau damping, following which, the hot electron is injected into the conduction band (CB) of the semiconductor. The electron-hole pairs generated in such a manner display a broad distribution of energies. (b) The IFCT mechanism where an electron in the noble metal is directly excited into the CB of the semiconductor, and its plasmonic counterpart in (c) PICTT where the plasmon dephases with the direct creation of an electron in the CB of the semiconductor and a hole in the metal. VB indicates the semiconductor valence band, while hν is the energy of the incident photon. Reprinted with permission from Ref [134] Copyright The American Association for the Advancement of Science (2015).
Surprisingly, multiple experimental observations have reported that the presence of a chemical adsorbate on a plasmonic nanoparticle can indeed lead to even faster relaxation of the LSPR over timescales of ~5 fs [123]. This relaxation of the LSPR induced by the chemical adsorbate is generally referred to as chemical interface damping (CID). CID describes how the addition of adsorbates, absent in the IFCT mechanism, to the surfaces of plasmonic nanostructures induces a broadening of the plasmon band, while providing another direct and additional pathway for the dephasing of the plasmon. Irrespective of the conflicting timescales for electron transfer from metal nanoparticles to a chemical adsorbate and the opposite case alike, excitation of an interfacial charge-transfer transition has still been observed. The difficulties in reconciling this new ultrafast relaxation time with the indirect sequential mechanism described in the conventional theory has led to the development of a contending explanation where molecule plasmon-enhanced photocatalysis reactions are suggested to proceed through a more direct mechanism or a “dissociation induced by electronic transitions” (DIET) process (Figure 5b) [61][117][123][144]. In this direct excitation mechanism, a charge-transfer transition is directly excited such that the prior processes of internal relaxation of electrons and damping of the LSPR within the metal nanoparticle are seemingly irrelevant. Furthermore, the DIET process has been used to explain the high quantum yields for hot electron charge transfer observed in plasmon-induced oxidation reactions involving resonant photo-induced electron transfer from Au and Ag nanoparticles to strongly bound molecules or semiconductor quantum dots [61][117][144]. DIET is a subset of CID describing small molecule plasmon-enhanced interfacial charge transfer processes where the excitation of the charge transfer transition transiently occupies a surface bound anionic state of the adsorbed molecule. Conventional theory describes this state to be vibrationally excited, and to relax rapidly through vibrational cooling followed by electron transfer back to the metal, but when the excitation rate overcomes the relaxation rate, dissociation can be activated resulting in reactive species that promote photocatalytic reactions [61]. The distinction between CID and electron transfer following plasmon decay remains an unsolved mystery. It is also unclear if CID and electron-surface scattering are distinct phenomena [145]. In the context of the present review, DIET is highly relevant to those cases where an organic molecule is adsorbed on the noble metal–TiO2 heterojunction such as: the oxidative degradation of organic compounds using TiO2–Au photocatalysts [70], the vapor phase reduction of CO2 over TiO2–Au NP photocatalysts [146], the photocatalytic oxidation of CO over Au NP–TiO2 [147], and the use of reduced graphene oxide (rGO) bridges to shuttle hot electrons more effectively between Ag/Au NPs and TiO2 [148][149].
A fundamental difference between the direct and sequential mechanisms revolves around the question of when plasmon-dephasing exactly occurs. The sequential mechanism describes plasmon dephasing to occur before electron-transfer reactions, whereas in the direct transfer mechanism, it is the charge-transfer reaction itself that leads to plasmon dephasing. The importance of this distinction between the two processes has signified the necessity for further research on CID processes to examine how molecules interact with electrons in metal nanoparticles as well as the need to identify the source and exact microscopic origin of the plasmon-induced hot electrons. Lee et al. [150] have explored possibilities towards controlling, tuning, and optimizing the contributions provided by CID as a channel for plasmonic hot-electron energy transfer. Using scanning electron microscopy-correlated dark field scattering, Lee et al. [150] studied the electronic nature of CID behavior for systems of benzene adsorbates on gold bipyramids with similar aspect ratios to those of gold nanorods. The bipyramidal morphology of the gold nanostructures alongside the electronic effects of the adsorbate molecules were observed to generate increased interfacial contact between the plasmonic noble metal and molecular adsorbate systems. As such, electron withdrawing groups on the adsorbates were found to induce larger homogeneous and high quality LSPR line widths as opposed to those of electron donating groups that experienced a weakened LSPR response due to back transfer of electrons. Lee et al. [150] demonstrated that CID in the LSPR can thus be tuned by controlling the electron withdrawing and electron donating features of the molecular adsorbates deposited on the surface of a plasmonic noble metal. Using a complementary approach, Foerster et al. [145] have demonstrated that CID scales inversely with the effective path length of electrons, in other words, the average distance of electrons to reach the surface, in the plasmonic noble metal. As such, they pose a resultant study demonstrating that by modifying the characteristic dimensions of the noble metal, i.e., gold nanorods of different sizes (14 × 41 nm, 18 × 55 nm, 22 × 66 nm, 27 × 78 nm) but similar aspect ratios, one can moderate the contribution of CID in comparison to other competing plasmon decay channels with CID becoming the dominating plasmon energy decay mechanism, in their work, via the observation of increased plasmon line width broadening for very small gold nanorods.
In the context of plasmonic photocatalysis, the sequential mechanism and the IFCT mechanism are used to describe and postulate the charge transfer mechanisms involved in plasmonic noble metal–semiconductor heterojunctions. Au and Ag are the preferred candidates as plasmonic noble metals, while TiO2 serves as the electron-accepting n-type semiconductor [151]. As shown in Figure 2, visible light absorption by the Au/Ag nanoparticle results in collective oscillations of sp band electrons, and the creation of hot electrons in the sp conduction band. These hot electrons dephase rapidly resulting in poor hot electron injection efficiencies. In order for successful hot electron injection to occur, the hot electrons as well as the resultant hot holes must satisfy a number of conditions: the charges must be able to reach the surface of the plasmonic nanoparticle, and from there have enough energy (above the metal’s Fermi level) to overcome the Schottky barrier, while the residual hot holes must be extracted to maintain charge neutrality within the plasmonic noble metal [152]. The charge separation process must conserve both the energy and momentum of carriers. The hot holes and electrons formed by this charge separation at the metal–semiconductor interface help initiate oxidation and reduction reactions, thus promoting plasmonic photocatalysis. Moving forward, it is necessary to combine the knowledge aggregated from the postulated theories as well as existing experiments to further probe and improve our understanding of hot electron phenomena so that we may harness the advantages they provide for plasmonic photocatalysis applications.
References
- Kalyanasundaram, K.; Graetzel, M. Artificial photosynthesis: Biomimetic approaches to solar energy conversion and storage. Curr. Opin. Biotechnol. 2010, 21, 298–310.
- BP. BP Statistical Review of World Energy; BP: London, UK, 2018.
- International Energy Agency. Key World Energy Statistics; International Energy Agency: Paris, France, 2018.
- U.S. Energy Information Administration. International Energy Outlook; U.S. Energy Information Administration: Washington, DC, USA, 2010.
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward solar fuels: Photocatalytic conversion of carbon dioxide to hydrocarbons. ACS Nano 2010, 4, 1259–1278.
- Wang, W.-N. Comparison of CO2 Photoreduction Systems: A Review. Aerosol Air Qual. Res. 2014.
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar Energy Supply and Storage for the Legacy and Nonlegacy Worlds. Chem. Rev. 2010, 110, 6474–6502.
- Hammarström, L. Overview: Capturing the Sun for Energy Production. AMBIO 2012, 41, 103–107.
- Li, J.; Wu, N. Semiconductor-based photocatalysts and photoelectrochemical cells for solar fuel generation: A review. Catal. Sci. Technol. 2015, 5, 1360–1384.
- Zhang, B.; Sun, L. Artificial photosynthesis: Opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264.
- Karthikeyan, C.; Arunachalam, P.; Ramachandran, K.; Al-Mayouf, A.M.; Karuppuchamy, S. Recent advances in semiconductor metal oxides with enhanced methods for solar photocatalytic applications. J. Alloys Compd. 2020, 828, 15.
- Zeng, S.; Kar, P.; Thakur, U.K.; Shankar, K. A review on photocatalytic CO2 reduction using perovskite oxide nanomaterials. Nanotechnology 2018, 29, 052001.
- Kumar, P.; Thakur, U.K.; Alam, K.; Kar, P.; Kisslinger, R.; Zeng, S.; Patel, S.; Shankar, K. Arrays of TiO2 nanorods embedded with fluorine doped carbon nitride quantum dots (CNFQDs) for visible light driven water splitting. Carbon 2018, 137, 174–187.
- Enesca, A.; Isac, L. Tandem Structures Semiconductors Based on TiO2_SnO2 and ZnO_SnO2 for Photocatalytic Organic Pollutant Removal. Nanomaterials 2021, 11, 200.
- Chandrasekaran, S.; Yao, L.; Deng, L.B.; Bowen, C.; Zhang, Y.; Chen, S.M.; Lin, Z.Q.; Peng, F.; Zhang, P.X. Recent advances in metal sulfides: From controlled fabrication to electrocatalytic, photocatalytic and photoelectrochemical water splitting and beyond. Chem. Soc. Rev. 2019, 48, 4178–4280.
- Kar, P.; Farsinezhad, S.; Zhang, X.; Shankar, K. Anodic Cu2S and CuS nanorod and nanowall arrays: Preparation, properties and application in CO2 photoreduction. Nanoscale 2014, 6, 14305–14318.
- Varadhan, P.; Fu, H.C.; Priante, D.; Retamal, J.R.D.; Zhao, C.; Ebaid, M.; Ng, T.K.; Ajia, I.; Mitra, S.; Roqan, I.S.; et al. Surface Passivation of GaN Nanowires for Enhanced Photoelectrochemical Water-Splitting. Nano Lett. 2017, 17, 1520–1528.
- Ebaid, M.; Priante, D.; Liu, G.Y.; Zhao, C.; Alias, M.S.; Buttner, U.; Ng, T.K.; Isimjan, T.T.; Idriss, H.; Ooi, B.S. Unbiased photocatalytic hydrogen generation from pure water on stable Ir-treated In0.33Ga0.67N nanorods. Nano Energy 2017, 37, 158–167.
- Wang, S.Y.; Hai, X.; Ding, X.; Chang, K.; Xiang, Y.G.; Meng, X.G.; Yang, Z.X.; Chen, H.; Ye, J.H. Light-Switchable Oxygen Vacancies in Ultrafine Bi5O7Br Nanotubes for Boosting Solar-Driven Nitrogen Fixation in Pure Water. Adv. Mater. 2017, 29, 7.
- Alam, K.M.; Kumar, P.; Kar, P.; Thakur, U.K.; Zeng, S.; Cui, K.; Shankar, K. Enhanced charge separation in g-C3N4–BiOI heterostructures for visible light driven photoelectrochemical water splitting. Nanoscale Adv. 2019, 1, 1460–1471.
- Farsinezhad, S.; Shanavas, T.; Mahdi, N.; Askar, A.M.; Kar, P.; Sharma, H.; Shankar, K. Core–shell titanium dioxide–titanium nitride nanotube arrays with near-infrared plasmon resonances. Nanotechnology 2018, 29, 154006.
- Reddy, K.R.; Reddy, C.V.; Nadagouda, M.N.; Shetti, N.P.; Jaesool, S.; Aminabhavi, T.M. Polymeric graphitic carbon nitride (g-C3N4)-based semiconducting nanostructured materials: Synthesis methods, properties and photocatalytic applications. J. Environ. Manag. 2019, 238, 25–40.
- Liu, Q.; Shi, J.; Xu, Z.Z.; Zhang, B.L.; Liu, H.L.; Lin, Y.L.; Gao, F.L.; Li, S.T.; Li, G.Q. InGaN Nanorods Decorated with Au Nanoparticles for Enhanced Water Splitting Based on Surface Plasmon Resonance Effects. Nanomaterials 2020, 10, 912.
- Dudita, M.; Bogatu, C.; Enesca, A.; Duta, A. The influence of the additives composition and concentration on the properties of SnOx thin films used in photocatalysis. Mater. Lett. 2011, 65, 2185–2189.
- Mouchaal, Y.; Enesca, A.; Mihoreanu, C.; Khelil, A.; Duta, A. Tuning the opto-electrical properties of SnO2 thin films by Ag+1 and In+3 co-doping. Mater. Sci. Eng. B 2015, 199, 22–29.
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 Photocatalysis: A Historical Overview and Future Prospects. Jpn. J. Appl. Phys. 2005, 44, 8269–8285.
- Ollis, D. Photocatalytic purification and remediation of contaminated air and water. Comptes Rendus l’Académie Sci. Ser. IIC Chem. 2000, 3, 405–411.
- Herrmann, J.-M. Heterogeneous photocatalysis: Fundamentals and applications to the removal of various types of aqueous pollutants. Catal. Today 1999, 53, 115–129.
- Indrakanti, V.P.; Kubicki, J.D.; Schobert, H.H. Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: Current state, chemical physics-based insights and outlook. Energy Environ. Sci. 2009, 2, 745–758.
- Wang, M.; Ye, M.; Iocozzia, J.; Lin, C.; Lin, Z. Plasmon-Mediated Solar Energy Conversion via Photocatalysis in Noble Metal/Semiconductor Composites. Adv. Sci. (Weinh.) 2016, 3, 1600024.
- Teixeira, I.F.; Homsi, M.S.; Geonmonond, R.S.; Rocha, G.; Peng, Y.K.; Silva, I.F.; Quiroz, J.; Camargo, P.H.C. Hot Electrons, Hot Holes, or Both? Tandem Synthesis of Imines Driven by the Plasmonic Excitation in Au/CeO2Nanorods. Nanomaterials 2020, 10, 1530.
- Hamans, R.F.; Kamarudheen, R.; Baldi, A. Single Particle Approaches to Plasmon-Driven Catalysis. Nanomaterials 2020, 10, 2377.
- Tran, V.T.; Nguyen, H.Q.; Kim, Y.M.; Ok, G.; Lee, J. Photonic-Plasmonic Nanostructures for Solar Energy Utilization and Emerging Biosensors. Nanomaterials 2020, 10, 2248.
- Fu, Y.S.; Li, J.; Li, J.G. Metal/Semiconductor Nanocomposites for Photocatalysis: Fundamentals, Structures, Applications and Properties. Nanomaterials 2019, 9, 359.
- Abed, J.; Rajput, N.S.; El Moutaouakil, A.; Jouiad, M. Recent Advances in the Design of Plasmonic Au/TiO2 Nanostructures for Enhanced Photocatalytic Water Splitting. Nanomaterials 2020, 10, 2260.
- Shibuta, M.; Yamamoto, K.; Ohta, T.; Inoue, T.; Mizoguchi, K.; Nakaya, M.; Eguchi, T.; Nakajima, A. Confined Hot Electron Relaxation at the Molecular Heterointerface of the Size-Selected Plasmonic Noble Metal Nanocluster and Layered C60. ACS Nano 2021, 15, 1199–1209.
- Park, Y.; Choi, J.; Kang, M.; Lee, H.; Ihee, H.; Park, J.Y. Relaxation Dynamics of Enhanced Hot-Electron Flow on Perovskite-Coupled Plasmonic Silver Schottky Nanodiodes. J. Phys. Chem. C 2021, 125, 2575–2582.
- Hattori, Y.; Meng, J.; Zheng, K.; Meier de Andrade, A.; Kullgren, J.; Broqvist, P.; Nordlander, P.; Sá, J. Phonon-Assisted Hot Carrier Generation in Plasmonic Semiconductor Systems. Nano Lett. 2021, 21, 1083–1089.
- Yang, W.; Liu, Y.; Cullen, D.A.; McBride, J.R.; Lian, T. Harvesting Sub-Bandgap IR Photons by Photothermionic Hot Electron Transfer in a Plasmonic p–n Junction. Nano Lett. 2021.
- Wang, A.; Wu, S.; Dong, J.; Wang, R.; Wang, J.; Zhang, J.; Zhong, S.; Bai, S. Interfacial facet engineering on the Schottky barrier between plasmonic Au and TiO2 in boosting the photocatalytic CO2 reduction under ultraviolet and visible light irradiation. Chem. Eng. J. (Lausanne) 2021, 404, 127145.
- Manuel, A.P.; Kirkey, A.; Mahdi, N.; Shankar, K. Plexcitonics—fundamental principles and optoelectronic applications. J. Mater. Chem. C 2019, 7, 1821–1853.
- Zeng, S.; Vahidzadeh, E.; VanEssen, C.G.; Kar, P.; Kisslinger, R.; Goswami, A.; Zhang, Y.; Mahdi, N.; Riddell, S.; Kobryn, A.E.; et al. Optical control of selectivity of high rate CO2 photoreduction via interband- or hot electron Z-scheme reaction pathways in Au-TiO2 plasmonic photonic crystal photocatalyst. Appl. Catal. B-Environ. 2020, 267, 118644.
- Ridley, B.K. Hot electrons in semiconductors. Sci. Prog. 1933 1986, 70, 425–459.
- Keyling, R.; Schöne, W.-D.; Ekardt, W. Comparison of the lifetime of excited electrons in noble metals. Phys. Rev. B 2000, 61, 1670–1673.
- Mukherjee, S.; Libisch, F.; Large, N.; Neumann, O.; Brown, L.V.; Cheng, J.; Lassiter, J.B.; Carter, E.A.; Nordlander, P.; Halas, N.J. Hot Electrons Do the Impossible: Plasmon-Induced Dissociation of H2 on Au. Nano Lett. 2013, 13, 240–247.
- Brongersma, M.L.; Halas, N.J.; Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 2015, 10, 25–34.
- Manuel, A.P.; Barya, P.; Riddell, S.; Zeng, S.; Alam, K.M.; Shankar, K. Plasmonic photocatalysis and SERS sensing using ellipsometrically modeled Ag nanoisland substrates. Nanotechnology 2020, 31, 365301.
- Karaballi, R.A.; Esfahani Monfared, Y.; Dasog, M. Photothermal Transduction Efficiencies of Plasmonic Group 4 Metal Nitride Nanocrystals. Langmuir 2020, 36, 5058–5064.
- Rej, S.; Mascaretti, L.; Santiago, E.Y.; Tomanec, O.; Kment, Š.; Wang, Z.; Zbořil, R.; Fornasiero, P.; Govorov, A.O.; Naldoni, A. Determining Plasmonic Hot Electrons and Photothermal Effects during H2 Evolution with TiN–Pt Nanohybrids. ACS Catal. 2020, 10, 5261–5271.
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37.
- Moskovits, M. Surface-enhanced spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826.
- Frischkorn, C.; Wolf, M. Femtochemistry at Metal Surfaces: Nonadiabatic Reaction Dynamics. Chem. Rev. 2006, 106, 4207–4233.
- Buntin, S.A.; Richter, L.J.; Cavanagh, R.R.; King, D.S. Optically Driven Surface Reactions: Evidence for the Role of Hot Electrons. Phys. Rev. Lett. 1988, 61, 1321–1324.
- Bonn, M.; Funk, S.; Hess, C.; Denzler, D.N.; Stampfl, C.; Scheffler, M.; Wolf, M.; Ertl, G. Phonon- Versus Electron-Mediated Desorption and Oxidation of CO on Ru(0001). Science 1999, 285, 1042.
- Kao, F.J.; Busch, D.G.; Gomes da Costa, D.; Ho, W. Femtosecond versus nanosecond surface photochemistry: O2+CO on Pt(111) at 80 K. Phys. Rev. Lett. 1993, 70, 4098–4101.
- Eustis, S.; El-Sayed, M.A. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem. Soc. Rev. 2006, 35, 209–217.
- Yu, X.; Liu, F.; Bi, J.; Wang, B.; Yang, S. Improving the plasmonic efficiency of the Au nanorod-semiconductor photocatalysis toward water reduction by constructing a unique hot-dog nanostructure. Nano Energy 2017, 33, 469–475.
- Wu, L.; Tsunenari, N.; Nishi, H.; Sugawa, K.; Otsuki, J.; Tatsuma, T. Two-Dimensional Arrays of Au Halfshells with Different Sizes for Plasmon-Induced Charge Separation. ChemistrySelect 2017, 2, 3744–3749.
- Wu, X.; Ming, T.; Wang, X.; Wang, P.; Wang, J.; Chen, J. High-Photoluminescence-Yield Gold Nanocubes: For Cell Imaging and Photothermal Therapy. ACS Nano 2010, 4, 113–120.
- Lou, Z.; Fujitsuka, M.; Majima, T. Pt–Au Triangular Nanoprisms with Strong Dipole Plasmon Resonance for Hydrogen Generation Studied by Single-Particle Spectroscopy. ACS Nano 2016, 10, 6299–6305.
- Hartland, G.V.; Besteiro, L.V.; Johns, P.; Govorov, A.O. What’s so Hot about Electrons in Metal Nanoparticles? ACS Energy Lett. 2017, 2, 1641–1653.
- Haegel, N. Relaxation semiconductors: In theory and in practice. Appl. Phys. A 1991, 53, 1–7.
- Zarifi, M.H.; Mohammadpour, A.; Farsinezhad, S.; Wiltshire, B.D.; Nosrati, M.; Askar, A.M.; Daneshmand, M.; Shankar, K. Time-Resolved Microwave Photoconductivity (TRMC) Using Planar Microwave Resonators: Application to the Study of Long-Lived Charge Pairs in Photoexcited Titania Nanotube Arrays. J. Phys. Chem. C 2015, 119, 14358–14365.
- DuChene, J.S.; Sweeny, B.C.; Johnston-Peck, A.C.; Su, D.; Stach, E.A.; Wei, W.D. Prolonged Hot Electron Dynamics in Plasmonic-Metal/Semiconductor Heterostructures with Implications for Solar Photocatalysis. Angew. Chem. Int. Ed. 2014, 53, 7887–7891.
- Sundararaman, R.; Narang, P.; Jermyn, A.S.; Goddard Iii, W.A.; Atwater, H.A. Theoretical predictions for hot-carrier generation from surface plasmon decay. Nat. Commun. 2014, 5, 5788.
- Brown, A.M.; Sundararaman, R.; Narang, P.; Goddard, W.A.; Atwater, H.A. Nonradiative Plasmon Decay and Hot Carrier Dynamics: Effects of Phonons, Surfaces, and Geometry. ACS Nano 2016, 10, 957–966.
- Hazra, A.; Das, S.; Kanungo, J.; Sarkar, C.K.; Basu, S. Studies on a resistive gas sensor based on sol–gel grown nanocrystalline p-TiO2 thin film for fast hydrogen detection. Sens. Actuators B Chem. 2013, 183, 87–95.
- Kar, P.; Zhang, Y.; Farsinezhad, S.; Mohammadpour, A.; Wiltshire, B.D.; Sharma, H.; Shankar, K. Rutile phase n-and p-type anodic titania nanotube arrays with square-shaped pore morphologies. Chem. Commun. 2015, 51, 7816–7819.
- Zhang, Y.; Zhou, S.; Su, X.; Xu, J.; Nie, G.; Zhang, Y.; He, Y.; Yu, S. Synthesis and characterization of Ag-loaded p-type TiO2 for adsorption and photocatalytic degradation of tetrabromobisphenol A. Water Environ. Res. 2020, 92, 713–721.
- Karbalaei Akbari, M.; Hai, Z.; Wei, Z.; Hu, J.; Zhuiykov, S. Wafer-scale two-dimensional Au-TiO2 bilayer films for photocatalytic degradation of Palmitic acid under UV and visible light illumination. Mater. Res. Bull. 2017, 95, 380–391.
- Jellison, G.E.; Boatner, L.A.; Budai, J.D.; Jeong, B.S.; Norton, D.P. Spectroscopic ellipsometry of thin film and bulk anatase (TiO2). J. Appl. Phys. 2003, 93, 9537–9541.
- Takahashi, M.; Tsukigi, K.; Uchino, T.; Yoko, T. Enhanced photocurrent in thin film TiO2 electrodes prepared by sol–gel method. Thin Solid Films 2001, 388, 231–236.
- Salvador, P. Hole diffusion length in n-TiO2 single crystals and sintered electrodes: Photoelectrochemical determination and comparative analysis. J. Appl. Phys. 1984, 55, 2977–2985.
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637.
- Wang, D.; Zhang, X.; Sun, P.; Lu, S.; Wang, L.; Wang, C.; Liu, Y. Photoelectrochemical Water Splitting with Rutile TiO2 Nanowires Array: Synergistic Effect of Hydrogen Treatment and Surface Modification with Anatase Nanoparticles. Electrochim. Acta 2014, 130, 290–295.
- Mohammadpour, A.; Kar, P.; Wiltshire, B.D.; Askar, A.M.; Shankar, K. Electron transport, trapping and recombination in anodic TiO2 nanotube arrays. Curr. Nanosci. 2015, 11, 593–614.
- Liu, L.; Zhao, H.; Andino, J.M.; Li, Y. Photocatalytic CO2 Reduction with H2O on TiO2 Nanocrystals: Comparison of Anatase, Rutile, and Brookite Polymorphs and Exploration of Surface Chemistry. ACS Catal. 2012, 2, 1817–1828.
- Pan, J.; Wu, X.; Wang, L.; Liu, G.; Lu, G.Q.; Cheng, H.-M. Synthesis of anatase TiO2 rods with dominant reactive facets for the photoreduction of CO2 to CH4 and use in dye-sensitized solar cells. Chem. Commun. 2011, 47, 8361–8363.
- Zhao, H.; Liu, L.; Andino, J.M.; Li, Y. Bicrystalline TiO2 with controllable anatase-brookite phase content for enhanced CO2 photoreduction to fuels. J. Mater. Chem. A 2013, 1, 8209–8216.
- Anpo, M.; Nakaya, H.; Kodama, S.; Kubokawa, Y.; Domen, K.; Onishi, T. Photocatalysis over binary metal oxides. Enhancement of the photocatalytic activity of titanium dioxide in titanium-silicon oxides. J. Phys. Chem. 1986, 90, 1633–1636.
- Wang, W.-N.; Park, J.; Biswas, P. Rapid synthesis of nanostructured Cu-TiO2-SiO2 composites for CO2 photoreduction by evaporation driven self-assembly. Catal. Sci. Technol. 2011, 1, 593–600.
- Kongkanand, A.; Tvrdy, K.; Takechi, K.; Kuno, M.; Kamat, P.V. Quantum Dot Solar Cells. Tuning Photoresponse through Size and Shape Control of CdSe−TiO2 Architecture. J. Am. Chem. Soc. 2008, 130, 4007–4015.
- Prabakar, K.; Minkyu, S.; Inyoung, S.; Heeje, K. CdSe quantum dots co-sensitized TiO 2 photoelectrodes: Particle size dependent properties. J. Phys. D Appl. Phys. 2010, 43, 012002.
- Sachs, M.; Pastor, E.; Kafizas, A.; Durrant, J.R. Evaluation of Surface State Mediated Charge Recombination in Anatase and Rutile TiO2. J. Phys. Chem. Lett. 2016, 7, 3742–3746.
- Li, H.; Bian, Z.; Zhu, J.; Huo, Y.; Li, H.; Lu, Y. Mesoporous Au/TiO2 Nanocomposites with Enhanced Photocatalytic Activity. J. Am. Chem. Soc. 2007, 129, 4538–4539.
- Zhou, M.; Bao, J.; Liang, L.; Xie, Y.; Wu, H.B.; Lou, X.W. Ordered macroporous BiVO4 architectures with controllable dual porosity for efficient solar water splitting. Angew. Chem. Int. Ed. 2013, 52, 8579–8583.
- Tian, Y.; Tatsuma, T. Mechanisms and applications of plasmon-induced charge separation at TiO2 films loaded with gold nanoparticles. J. Am. Chem. Soc. 2005, 127, 7632–7637.
- Radzig, M.; Koksharova, O.; Khmel, I.; Ivanov, V.; Yorov, K.; Kiwi, J.; Rtimi, S.; Tastekova, E.; Aybush, A.; Nadtochenko, V. Femtosecond Spectroscopy of Au Hot-Electron Injection into TiO2: Evidence for Au/TiO2 Plasmon Photocatalysis by Bactericidal Au Ions and Related Phenomena. Nanomaterials 2019, 9, 217.
- Negrin-Montecelo, Y.; Testa-Anta, M.; Marin-Caba, L.; Perez-Lorenzo, M.; Salgueirino, V.; Correa-Duarte, M.A.; Comesana-Hermo, M. Titanate Nanowires as One-Dimensional Hot Spot Generators for Broadband Au-TiO2 Photocatalysis. Nanomaterials 2019, 9, 990.
- Linh, V.T.N.; Xiao, X.F.; Jung, H.S.; Giannini, V.; Maier, S.A.; Kim, D.H.; Lee, Y.I.; Park, S.G. Compact Integration of TiO2 Nanoparticles into the Cross-Points of 3D Vertically Stacked Ag Nanowires for Plasmon-Enhanced Photocatalysis. Nanomaterials 2019, 9, 468.
- White, T.P.; Catchpole, K.R. Plasmon-enhanced internal photoemission for photovoltaics: Theoretical efficiency limits. Appl. Phys. Lett. 2012, 101, 073905.
- Yu, H.; Peng, Y.; Yang, Y.; Li, Z.-Y. Plasmon-enhanced light–matter interactions and applications. NPJ Comput. Mater. 2019, 5, 45.
- Zorić, I.; Zäch, M.; Kasemo, B.; Langhammer, C. Gold, Platinum, and Aluminum Nanodisk Plasmons: Material Independence, Subradiance, and Damping Mechanisms. ACS Nano 2011, 5, 2535–2546.
- Vahidzadeh, E.; Zeng, S.; Manuel, A.P.; Riddell, S.; Kumar, P.; Alam, K.M.; Shankar, K. Asymmetric Multipole Plasmon-Mediated Catalysis Shifts the Product Selectivity of CO2 Photoreduction toward C2+ Products. ACS Appl. Mater. Inter. 2021, 13, 7248–7258.
- Lee, K.-S.; El-Sayed, M.A. Gold and Silver Nanoparticles in Sensing and Imaging: Sensitivity of Plasmon Response to Size, Shape, and Metal Composition. J. Phys. Chem. B 2006, 110, 19220–19225.
- Link, S.; Wang, Z.L.; El-Sayed, M.A. Alloy Formation of Gold−Silver Nanoparticles and the Dependence of the Plasmon Absorption on Their Composition. J. Phys. Chem. B 1999, 103, 3529–3533.
- Jiang, T.; Jia, C.; Zhang, L.; He, S.; Sang, Y.; Li, H.; Li, Y.; Xu, X.; Liu, H. Gold and gold-palladium alloy nanoparticles on heterostructured TiO2 nanobelts as plasmonic photocatalysts for benzyl alcohol oxidation. Nanoscale 2015, 7, 209–217.
- Wiley, B.J.; Im, S.H.; Li, Z.-Y.; McLellan, J.; Siekkinen, A.; Xia, Y. Maneuvering the Surface Plasmon Resonance of Silver Nanostructures through Shape-Controlled Synthesis. J. Phys. Chem. B 2006, 110, 15666–15675.
- Bonatti, L.; Gil, G.; Giovannini, T.; Corni, S.; Cappelli, C. Plasmonic Resonances of Metal Nanoparticles: Atomistic vs. Continuum Approaches. Front. Chem. 2020, 8.
- Gutiérrez, Y.; Brown, A.S.; Moreno, F.; Losurdo, M. Plasmonics beyond noble metals: Exploiting phase and compositional changes for manipulating plasmonic performance. J. Appl. Phys. 2020, 128, 080901.
- Zhao, Y.; Burda, C. Development of plasmonic semiconductor nanomaterials with copper chalcogenides for a future with sustainable energy materials. Energy Environ. Sci. 2012, 5, 5564–5576.
- Naik, G.V.; Schroeder, J.L.; Ni, X.; Kildishev, A.V.; Sands, T.D.; Boltasseva, A. Titanium nitride as a plasmonic material for visible and near-infrared wavelengths. Opt. Mater. Express 2012, 2, 478–489.
- Karaballi, R.A.; Humagain, G.; Fleischman, B.R.; Dasog, M. Synthesis of Plasmonic Group-4 Nitride Nanocrystals by Solid-State Metathesis. Angew. Chem. 2019, 131, 3179–3182.
- Huang, X.; El-Sayed, I.H.; Qian, W.; El-Sayed, M.A. Cancer Cell Imaging and Photothermal Therapy in the Near-Infrared Region by Using Gold Nanorods. J. Am. Chem. Soc. 2006, 128, 2115–2120.
- Hirsch, L.R.; Stafford, R.J.; Bankson, J.A.; Sershen, S.R.; Rivera, B.; Price, R.E.; Hazle, J.D.; Halas, N.J.; West, J.L. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc. Natl. Acad. Sci. USA 2003, 100, 13549–13554.
- Laura, B.C.; Lissett, R.B.; Germaine, A.; Tse-Kuan, Y.; Rachel, S.; Yi, L.; Rebekah, A.D. Immunoconjugated gold nanoshell-mediated photothermal ablation of trastuzumab-resistant breast cancer cells. Breast Cancer Res. Treat. 2011, 125, 27–34.
- Zhou, W.; Dridi, M.; Suh, J.Y.; Kim, C.H.; Co, D.T.; Wasielewski, M.R.; Schatz, G.C.; Odom, T.W. Lasing action in strongly coupled plasmonic nanocavity arrays. Nat. Nanotechnol. 2013, 8, 506.
- Lu, Y.-J.; Kim, J.; Chen, H.-Y.; Wu, C.; Dabidian, N.; Sanders, C.E.; Wang, C.-Y.; Lu, M.-Y.; Li, B.-H.; Qiu, X.; et al. Plasmonic Nanolaser Using Epitaxially Grown Silver Film. Science 2012, 337, 450.
- Jain, P.K.; Huang, X.; El-Sayed, I.H.; El-Sayed, M.A. Noble Metals on the Nanoscale: Optical and Photothermal Properties and Some Applications in Imaging, Sensing, Biology, and Medicine. Acc. Chem. Res. 2008, 41, 1578–1586.
- Loo, C.; Lin, A.; Hirsch, L.; Lee, M.-H.; Barton, J.; Halas, N.; West, J.; Drezek, R. Nanoshell-Enabled Photonics-Based Imaging and Therapy of Cancer. Technol. Cancer Res. Treat. 2004, 3, 33–40.
- Moskovits, M. Surface-enhanced Raman spectroscopy: A brief retrospective. J. Raman Spectrosc. 2005, 36, 485–496.
- Marimuthu, A.; Christopher, P.; Linic, S. Design of Plasmonic Platforms for Selective Molecular Sensing Based on Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2012, 116, 9824–9829.
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011, 10, 911.
- Zhou, X.; Liu, G.; Yu, J.; Fan, W. Surface plasmon resonance-mediated photocatalysis by noble metal-based composites under visible light. J. Mater. Chem. 2012, 22, 21337–21354.
- Atwater, H.A.; Polman, A. Plasmonics for improved photovoltaic devices. Nat. Mater. 2010, 9, 205–213.
- Li, Y.; Yan, X.; Wu, Y.; Zhang, X.; Ren, X. Plasmon-Enhanced Light Absorption in GaAs Nanowire Array Solar Cells. Nanoscale Res. Lett. 2015, 10, 436.
- Boerigter, C.; Aslam, U.; Linic, S. Mechanism of Charge Transfer from Plasmonic Nanostructures to Chemically Attached Materials. ACS Nano 2016, 10, 6108–6115.
- Alexei, D.S.; Gregory, N.G.t.; Roman, S. Hot-electron effect in superconductors and its applications for radiation sensors. Supercond. Sci. Technol. 2002, 15, R1.
- Le Ru, E.C.; Etchegoin, P.G. Principles of Surface-Enhanced Raman Spectroscopy. [Electronic Resource]: And Related Plasmonic Effects, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2009.
- Jana, J.; Ganguly, M.; Pal, T. Enlightening surface plasmon resonance effect of metal nanoparticles for practical spectroscopic application. RSC Adv. 2016, 6, 86174–86211.
- Wark, A.W.; Lee, H.J.; Corn, R.M. Long-Range Surface Plasmon Resonance Imaging for Bioaffinity Sensors. Anal. Chem. 2005, 77, 3904–3907.
- Stewart, M.E.; Anderton, C.R.; Thompson, L.B.; Maria, J.; Gray, S.K.; Rogers, J.A.; Nuzzo, R.G. Nanostructured Plasmonic Sensors. Chem. Rev. 2008, 108, 494–521.
- Kale, M.J.; Avanesian, T.; Christopher, P. Direct Photocatalysis by Plasmonic Nanostructures. ACS Catal. 2013, 4, 116–128.
- Sönnichsen, C.; Franzl, T.; Wilk, T.; von Plessen, G.; Feldmann, J.; Wilson, O.; Mulvaney, P. Drastic Reduction of Plasmon Damping in Gold Nanorods. Phys. Rev. Lett. 2002, 88, 077402.
- Lamprecht, B.; Leitner, A.; Aussenegg, F.R. SHG studies of plasmon dephasing in nanoparticles. Appl. Phys. B 1999, 68, 419–423.
- Tsu, R. Landau Damping and Dispersion of Phonon, Plasmon, and Photon Waves in Polar Semiconductors. Phys. Rev. 1967, 164, 380–383.
- Perner, M.; Klar, T.; Grosse, S.; Lemmer, U.; von Plessen, G.; Spirkl, W.; Feldmann, J. Homogeneous line widths of surface plasmons in gold nanoparticles measured by femtosecond pump-and-probe and near-field optical spectroscopy. J. Lumin. 1998, 76–77, 181–184.
- Voisin, C.; Christofilos, D.; Del Fatti, N.; Vallée, F.; Prével, B.; Cottancin, E.; Lermé, J.; Pellarin, M.; Broyer, M. Size-Dependent Electron-Electron Interactions in Metal Nanoparticles. Phys. Rev. Lett. 2000, 85, 2200–2203.
- Voisin, C.; Del Fatti, N.; Christofilos, D.; Vallée, F. Ultrafast Electron Dynamics and Optical Nonlinearities in Metal Nanoparticles. J. Phys. Chem. B 2001, 105, 2264–2280.
- Ratchford, D.C.; Dunkelberger, A.D.; Owrutsky, J.C.; Pehrsson, P.E.; Vurgaftman, I. Quantification of Efficient Plasmonic Hot-Electron Injection in Gold Nanoparticle-TiO2 Films. Nano Lett. 2017, 17, 6047–6055.
- Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast Plasmon-Induced Electron Transfer from Gold Nanodots into TiO2 Nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853.
- Du, L.; Furube, A.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast plasmon induced electron injection mechanism in gold–TiO2 nanoparticle system. J. Photochem. Photobiol. C Photochem. Rev. 2013, 15, 21–30.
- Du, L.; Furube, A.; Yamamoto, K.; Hara, K.; Katoh, R.; Tachiya, M. Plasmon-Induced Charge Separation and Recombination Dynamics in Gold−TiO2 Nanoparticle Systems: Dependence on TiO2 Particle Size. J. Phys. Chem. C 2009, 113, 6454–6462.
- Wu, K.; Chen, J.; McBride, J.R.; Lian, T. Efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition. Science 2015, 349, 632.
- Nosaka, Y.; Takahashi, S.; Sakamoto, H.; Nosaka, A.Y. Reaction Mechanism of Cu(II)-Grafted Visible-Light Responsive TiO2 and WO3 Photocatalysts Studied by Means of ESR Spectroscopy and Chemiluminescence Photometry. J. Phys. Chem. C 2011, 115, 21283–21290.
- Nogawa, T.; Isobe, T.; Matsushita, S.; Nakajima, A. Preparation and visible-light photocatalytic activity of Au- and Cu-modified TiO2 powders. Mater. Lett. 2012, 82, 174–177.
- Zhang, X.; Han, F.; Shi, B.; Farsinezhad, S.; Dechaine, G.P.; Shankar, K. Photocatalytic Conversion of Diluted CO2 into Light Hydrocarbons Using Periodically Modulated Multiwalled Nanotube Arrays. Angew. Chem. 2012, 124, 12904–12907.
- Farsinezhad, S.; Sharma, H.; Shankar, K. Interfacial band alignment for photocatalytic charge separation in TiO2 nanotube arrays coated with CuPt nanoparticles. Phys. Chem. Chem. Phys. 2015, 17, 29723–29733.
- Rtimi, S.; Pulgarin, C.; Sanjines, R.; Kiwi, J. Accelerated self-cleaning by Cu promoted semiconductor binary-oxides under low intensity sunlight irradiation. Appl. Catal. B-Environ. 2016, 180, 648–655.
- Bernareggi, M.; Dozzi, M.; Bettini, L.; Ferretti, A.; Chiarello, G.; Selli, E. Flame-Made Cu/TiO2 and Cu-Pt/TiO2 Photocatalysts for Hydrogen Production. Catalysts 2017, 7, 301.
- Kar, P.; Zhang, Y.; Mahdi, N.; Thakur, U.K.; Wiltshire, B.D.; Kisslinger, R.; Shankar, K. Heterojunctions of mixed phase TiO2 nanotubes with Cu, CuPt, and Pt nanoparticles: Interfacial band alignment and visible light photoelectrochemical activity. Nanotechnology 2017, 29, 014002.
- Asbury, J.B.; Hao, E.; Wang, Y.; Ghosh, H.N.; Lian, T. Ultrafast Electron Transfer Dynamics from Molecular Adsorbates to Semiconductor Nanocrystalline Thin Films. J. Phys. Chem. B 2001, 105, 4545–4557.
- Anderson, N.A.; Lian, T. Ultrafast Electron Transfer at the Molecule-Semiconductor Nanoparticle Interface. Annu. Rev. Phys. Chem. 2004, 56, 491–519.
- Boerigter, C.; Campana, R.; Morabito, M.; Linic, S. Evidence and implications of direct charge excitation as the dominant mechanism in plasmon-mediated photocatalysis. Nat. Commun. 2016, 7, 10545.
- Foerster, B.; Kaefer, K.; Celiksoy, S.; Sönnichsen, C.; Joplin, A.; Link, S. Chemical Interface Damping Depends on Electrons Reaching the Surface. ACS Nano 2017, 11, 2886–2893.
- Hou, W.; Hung, W.H.; Pavaskar, P.; Goeppert, A.; Aykol, M.; Cronin, S.B. Photocatalytic Conversion of CO2 to Hydrocarbon Fuels via Plasmon-Enhanced Absorption and Metallic Interband Transitions. ACS Catal. 2011, 1, 929–936.
- Deng, X.Q.; Zhu, B.; Li, X.S.; Liu, J.L.; Zhu, X.B.; Zhu, A.M. Visible-light photocatalytic oxidation of CO over plasmonic Au/TiO2: Unusual features of oxygen plasma activation. Appl. Catal. B-Environ. 2016, 188, 48–55.
- Subramanian, A.; Pan, Z.H.; Li, H.F.; Zhou, L.S.; Li, W.F.; Qiu, Y.C.; Xu, Y.J.; Hou, Y.; Muzi, C.; Zhang, Y.G. Synergistic promotion of photoelectrochemical water splitting efficiency of TiO2 nanorods using metal-semiconducting nanoparticles. Appl. Surf. Sci. 2017, 420, 631–637.
- Lang, Q.Q.; Chen, Y.H.; Huang, T.L.; Yang, L.N.; Zhong, S.X.; Wu, L.J.; Chen, J.R.; Bai, S. Graphene “bridge” in transferring hot electrons from plasmonic Ag nanocubes to TiO2 nanosheets for enhanced visible light photocatalytic hydrogen evolution. Appl. Catal. B-Environ. 2018, 220, 182–190.
- Lee, S.Y.; Tsalu, P.V.; Kim, G.W.; Seo, M.J.; Hong, J.W.; Ha, J.W. Tuning Chemical Interface Damping: Interfacial Electronic Effects of Adsorbate Molecules and Sharp Tips of Single Gold Bipyramids. Nano Lett. 2019, 19, 2568–2574.
- Shiraishi, Y.; Yasumoto, N.; Imai, J.; Sakamoto, H.; Tanaka, S.; Ichikawa, S.; Ohtani, B.; Hirai, T. Quantum tunneling injection of hot electrons in Au/TiO2 plasmonic photocatalysts. Nanoscale 2017, 9, 8349–8361.
- Valenti, M.; Venugopal, A.; Schmidt-Ott, A.; Smith, W.A.; Tordera, D.; Jonsson, M.P.; Biskos, G. Hot Carrier Generation and Extraction of Plasmonic Alloy Nanoparticles. ACS Photonics 2017, 4, 1146–1152.


