 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Camelia Diaconu | + 6859 word(s) | 6859 | 2021-04-25 12:11:36 | | | |
| 2 | Conner Chen | Meta information modification | 6859 | 2021-05-07 03:21:18 | | |
Video Upload Options
Non-alcoholic fatty liver disease (NAFLD) is a liver disease that can progress from hepatic steatosis to steatohepatitis and even cirrhosis and hepatocellular carcinoma. From the histopathological point of view, it is characterized by excess storage of macrovesicular fat in hepatocytes. These macrovesicular storages are composed of triglycerides that accumulate in the liver. The process can lead in some individuals to an inflammatory response, which is responsible for steatohepatitis, that leads to fibrosis and, finally, cirrhosis.NAFLD has a growing prevalence in recent years. Its association with cardiovascular disease has been intensively studied, and certain correlations have been identified. The connection between these two entities has lately aroused interest regarding therapeutic management.
1. Non-alcoholic fatty liver disease (NAFLD) and Cardiovascular Comorbidities
The risk factors for developing NAFLD are metabolic syndrome (central adiposity, hyperglycemia, dyslipidemia, arterial hypertension), weight gain, and insulin resistance/diabetes [1][2].
NAFLD can be diagnosed either by imaging techniques or histological examination. Both techniques should be supported by the exclusion of other causes of fatty liver (viral infections, alcohol consumption, autoimmunity, or certain drugs).
Regarding imaging techniques, abdominal ultrasonography is the most used because its feasibility and low costs [3][4]. Magnetic resonance imaging (MRI), that measures hepatic fat concentration, can also be used, but it is more expensive [5][6].
The standard diagnostic tool of NAFLD is hepatic biopsy. However, this is an invasive and a more expensive procedure and can also be fraught with sampling error and variability in pathologist interpretation [7]. The histological criteria for diagnosing steatohepatitis include the following:
-
>5% macrovesicular steatosis;
-
presence of inflammation;
-
ballooning hepatocytes with predominantly centro-lobular distribution [8].
Biological findings in NAFLD are high serum triglycerides and low levels of high-density lipoprotein cholesterol (HDL-C). Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) can also be mildly increased, with ALT levels higher than AST, but this is more specific to non-alcoholic steatohepatitis (NASH) [9].
The pooled prevalence of NAFLD globally is 25.24%. The highest prevalence rates have been reported in South and Middle East American countries (30%). Similar NAFLD prevalence has been reported in Europe (24%) [1]. Taking into consideration sex differences, the prevalence is higher in men than women but increases in women after menopause [10].
Traditionally, NAFLD was described as a hepatic manifestation of the metabolic syndrome. Recently, it has started to be recognized as a multisystemic disease, which is involved in the pathogenesis of various conditions, such as type 2 diabetes mellitus, cardiovascular disease, and chronic kidney disease [11]. Correlations between NAFLD and cardiovascular comorbidities, independent of traditional cardiovascular risk and metabolic syndrome (MetS), were also demonstrated. NAFLD is associated with hypertension, atherogenic dyslipidemia, that leads to chronic coronary syndrome and stroke, arrhythmias, increased risk of thromboembolic events, and structural heart disease.
2. Non-Alcoholic Fatty Liver Disease and Arterial Hypertension
Arterial hypertension affects nearly 30% of the adults and is a result of the combination between genetic predisposition and environmental risk factors [12]. Hypertension has long been demonstrated to be associated with NAFLD, studies revealing a high prevalence of NAFLD in hypertensive patients, even without the risk factors for liver steatosis [13]. Although numerous research data show that NAFLD is an independent risk factor for hypertension, all have limitations regarding the techniques used for NAFLD diagnosis, the type of study (e.g., cross-sectional), the use of relative small scales, or they miss some diseases from patients’ history, by gathering information from medical interviews [14][15][16][17]. Taking all this information into account, the question to be answered is what the pathogenic mechanisms behind this association are.
There are many theories that explain how NAFLD can induce hypertension, but most of them have been studied on experimental animals, and the extent of these that can be applied to humans needs further investigations. These theories are systemic inflammation, insulin resistance, increased vasoconstriction and decreased vasodilation, arterial stiffness, increased oxidative stress, gut dysbiosis, and genetic and epigenetic modifications.
2.1. Systemic Inflammation
Studies have shown that NAFLD strongly associates with increased levels of inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), and C-reactive protein (CRP) [18]. In addition, these cytokines are elevated in patients with primary hypertension [19].
Liver is an innate immune organ and the continuing production of metabolites, in this case triglycerides that accumulates in hepatocytes, leads to excessive inflammatory cytokines release, that promotes hepatic steatosis and fibrosis [12][20].
Other mechanisms that are involved in the activation of systemic inflammation are increased release of very-low-density lipoproteins (VLDL) from the overloaded triglyceride hepatocytes that stimulates Toll-like receptors (TLRs) and increased levels of hepatokines (fetuin A and retinol-binding protein 4) [21][22].
The innate immune components and TLRs are playing important roles in the induction and sustenance of hypertension.
Another mechanism that is involved in the development of hypertension is inflammation-induced activation of the renine-angiotensin system (RAS). TNF-α and IL-6 are responsible for the regulation of RAS components, especially angiotensinogen production in the liver and kidney, promoting angiotensin-related hypertension.
Moreover, various clinical evidence has demonstrated the correlation between NAFLD and chronic kidney disease, which is also a cause of hypertension.
In conclusion, these studies contribute to the theory that inflammation that appears in NAFLD is a cause of hypertension that accompanies this affliction [23].
2.2. Insulin Resistance
The mechanisms that are behind insulin resistance (IR) and hypertension are the activation of sympathetic nervous system and the renal sodium retention [24]. IR in NAFLD might be caused by a variety of factors, such as: hepatokines secretion, cytokines, and farnesoid X receptors (FXR).
Hepatokines originate from the fatty liver. They are able, through VLDL to alter the fatty acids metabolism. Moreover, they induce inflammation and insulin resistance in other cell types [25]. IR itself, is also responsible for the development of NAFLD [26]. Insulin is responsible for vasodilatation through nitric oxide (NO) production, and in reverse, IR stimulates vasoconstriction, leading also to hypertension.
FXR are identified as a bile acid activated nuclear receptors. They control lipid metabolism by regulating the synthesis, conjugation and transport of the bile acids. In NAFLD, their number is increased and they contribute to the suppression of hepatic FXR-mediated metabolic signaling, which further promotes IR [27].
All of the above considered, it can be stated that IR is, as most, a part of the mechanisms that induce hypertension in NAFLD patients, as it is one of the causes that leads to fatty liver disease.
2.3. Increased Vasoconstriction and Decreased Vasodilation
Asymmetric dimethylarginine (ADMA) is a recently discovered marker of endothelial dysfunction and atherosclerosis. It is generated by breaking down the proteins that were post-translationally methylated at an arginine residue [28]. ADMA is endogenously responsible for the inhibition of nitric oxide synthase (NOS), therefore causing the inhibition of vasodilatation. It has been observed that ADMA levels are significantly higher in patients with NAFLD (liver being the main site of elimination), even without traditional cardiovascular risk factors [28]. In addition, there is evidence that ADMA associates with endothelial dysfunction in patients with hypertension [29]. Several studies have shown that circulating ADMA is increased in subjects with histological-proven NAFLD: one study had only male subjects enrolled, with no morbid obesity, type 2 diabetes mellitus (T2DM), or hypertension, and another one showed that ADMA levels are higher in insulin resistance states [30] and that plasma ADMA levels are decreasing as a response to insulin sensitivity improvement [28][31]. Although several studies have demonstrated the correlation between vasoconstriction, ADMA and NAFLD, further investigation still needs to be done given the heterogeneity of the current studies.
2.4. Arterial Stiffness
Arterial stiffness results as a consequence of the complex interaction between stable and dynamic effects in cellular and structural components of the vascular wall [32]. The resistance and compliancy of the arteries are determined by two main structural components: elastin and collagen. Their quantity is kept stable by the interplay between production and degradation. Disruption of this balance, stimulated by an inflammatory context, might lead to reduced quantities of normal elastin and overproduction of altered collagen, therefore leading to increased arterial stiffness [33]. One theory that comes into the support of arterial stiffness and NAFLD is inflamed visceral adipose tissue. In this case, NASH might be responsible for releasing inflammatory, pro-thrombotic and oxidative-stress substances and NAFLD interferes with insulin resistance and atherogenic dyslipidemia [34][35][36].
Several studies, most of them with cross-sectional designs [37][38][39][40] confirmed the connection between increased arterial stiffness and NAFLD, independently of any additional cardio-metabolic predisposing factors. The weaknesses of these studies were their designs, that allow only mere correlations to be made and that the mainly diagnostic method used for NAFLD was ultrasonography. Ozturk et al. used biopsy for the diagnosis of NAFLD. They concluded that independently of MetS, NAFLD is responsible for increasing the risk of atherosclerosis and impaired function of the endothelium in adult male subjects [41].
In resume, the possible biological mechanisms that link NAFLD to increased arterial stiffness remain still unknown, but they possibly involve adipokines imbalance and chronic low-grade inflammation [32][42].
2.5. Increased Oxidative Stress
Oxidative stress reflects a disparity between the availability of reactive oxygen species (ROS) and cellular antioxidant system. This imbalance leads to the alteration of the cell functions and eventually cellular death [43]. These ROS are produced by the body due to normal intracellular metabolism and have physiological roles at low concentration, but, in high concentration, they can damage deoxyribonucleic acid (DNA) [44]. NAFLD has been suggested to be linked to oxidative stress [12][45].
One major source of oxidative stress is represented by homocysteine and the liver is a major metabolic organ for this. Alterations of the homocysteine metabolism involves impaired re-methylation to methionine and reduced trans sulphuration to cysteine. Therefore, intrahepatic vascular resistance is increased due to impaired NO formation, as homocysteine reduces NO release from sinusoidal endothelial cells and also causes hepatic stellate cell contraction [46]. Moreover, the production of ROS in NAFLD is responsible for the oxidation of low-density lipoproteins (LDL), which induces the transformation of the macrophages into foam cells. This is the first step of the atherosclerotic lesion development [47].
All these particularities described above suggest that oxidative stress is in charge for both increased intrahepatic resistance and atherosclerosis. In addition, clinical evidence has shown that elevated homocysteine levels are associated with hypertension [48].
2.6. Gut Dysbiosis
Axenic animal models have long been used to assess the repercussions that the absence of gut microbiota have.
Regarding NAFLD, it has been observed that the absenteeism of intestinal flora in mice leads to the gathering of active constituent ligands/androstane receptor (CAR), bile acids, bilirubin, and steroid hormones, which lead to alteration of hepatic xenobiotic metabolism, which could favor the development of NAFLD [49]. The mechanisms that are involved in the correlation between gut microbiota and NAFLD are changes in the quantity of energy absorbed from food, changes in the permeability of the intestine, and alterations of the expression of the genes involved in the lipogenesis. In addition, choline and bile acid metabolic signaling pathways are involved mechanisms, along with the production of ethanol in the intestine and interactions with the innate immunity [50].
On the other hand, gut microbiota and, specifically, dysbiosis has an important role in hypertension development [12]. Regarding the effect that dysbiosis has on hypertension, the following mechanisms are involved: inflammation, vasodilatation, and short-chain fatty acids. Intestinal dysbiosis regulates the differentiation and maturation of immune cells, therefore being implied in the systemic inflammatory state [51].
Vasodilatation is a mechanism that protects against hypertension. It seems to be caused by short-chain fatty acids in the presence of specific gut microbes, a process which is mediated through G-coupled protein receptors. The intestinal flora of the patients suffering from prehypertension and hypertension had a larger amount of Prevotella, a type of enteric bacteria, whereas the intestinal flora of controls individuals contained mostly bacteria from the genus Bacteroidetes [51].
So, it can be assumed that there is a correlation between hypertension, NAFLD, and gut dysbiosis.
2.7. Genetic and Epigenetic Modifications
Although there are consistent data that prove the link between hypertension and NAFLD, genetics research is limited regarding this association [12]. Family studies demonstrate that first degree relatives of the NAFLD patients have a much higher risk of developing the disease than the general population, and same familial distribution has long been proved for primary hypertension [52].
Adiponectin (ADIPOQ) gene (which encodes adiponectin) polymorphisms have been indicated to be the link between hypertension and NAFLD [53]. Adiponectin is a hormone derived from adipocytes. It has an important role in modulating lipid and glucose metabolism [54].
Another gene that was investigated was angiotensin receptor type 1 (AGTR1). In a recent prospective cohort study, the gain-of function A1166C (rs5186) variant in the AGTR1 gene represented a strong predictor for incident NAFLD and associated hypertension. In addition, polymorphism in AGTR1 may influence the risk of liver fibrosis in NAFLD [55].
Regarding the epigenetic changes, they are responsible for the interaction with inherited risk factors and, therefore, determine the susceptibility of NAFLD, hypertension, and cardiovascular disease [56][57].
Although promising research data show a clear link between hypertension and NAFLD, considering genetics and epigenetics, many data are still missing and more detailed investigations should be carried out.
3. Non-Alcoholic Fatty Liver Disease and Coronary Heart Disease
Several studies have proven the link between NAFLD and coronary artery disease (CAD). Most of them have demonstrated a correlation between hepatic steatosis and the calcification of coronary arteries [58][59].
In a meta-analysis, Ampuero et al. evaluated the impact of NAFLD on subclinical atherosclerosis and CAD. 14 studies were reviewed. CAD was taken into consideration when patients showed ≤50% stenosis in one or more of the major coronary arteries. Hepatic biopsy and ultrasound were used to evaluate NAFLD. Outcomes revealed that the intima-media thickness of carotid in patients with NAFLD had a higher prevalence (35.1% vs. 21.8%). On the other hand, four studies that assessed CAD by coronary angiogram showed that 80.4% of the subjects with NAFLD had CAD, while only 60.7% of the patients without NAFLD had CAD. The conclusion was that NAFLD increases the risk of coronary artery disease [60].
Another cross-sectional study, carried out by Chang et al., evaluated coronary artery calcification and its relationship with alcohol-induced fatty liver disease and (NAFLD). One hundred five thousand three hundred and twenty-eight Korean adults were included. Coronary artery calcium (CAC) score was assessed using computed tomography, liver fat by ultrasound and alcohol intake by g/day. The authors came to the conclusion that both alcohol-induced fatty liver disease and NAFLD were firmly associated with CAC score, without significant interaction with obesity [61].
Besides the fact that the relationship between coronary heart disease and NAFLD was strongly investigated, and that the correlation is clear, the pathophysiology and mechanisms that are behind still need to be investigated. Currently, it is thought to be a complex process that involves insulin resistance, adipokines, oxidative stress, and apoptosis [62].
3.1. Insulin Resistance
This theory is supported by various studies demonstrating that the higher the liver fat content is the lesser the hepatic insulin sensitivity is.
Kotronen et al. carried out a study where T2DM patients were examined. They concluded that insulin resistance in patients with increased liver fat is due to diminished insulin clearance. Sixty-eight patients with T2DM were compared to a control group containing non-diabetic subjects. Liver steatosis was determined by proton magnetic resonance spectroscopy. The clearance and action of insulin was assessed by the infusion of (3-3H) glucose and by the euglycemic insulin clamp technique. In addition, MRI was used in order to determine the body composition. The results confirmed that type 2 diabetic patients had higher liver fat (54%) and lower insulin clearance (24%) than nondiabetic subjects. In addition, their insulin levels were higher (34 mU/L vs. 25 mU/L) [63].
Jun N-terminal kinases (JNKs) are kinases involved in the survival and differentiation of the cells. They belong to the mitogen-activated protein kinase (MAPK) superfamily [64]. JNKs are involved in insulin resistance and have important roles regarding the β-cells of the pancreas. Those roles are concerning their secretory function and survival. They are activated by inflammatory cytokines. It has been found that insulin resistance is associated with the formation of autophagosomes in pancreatic β-cells [65]. The main mechanisms that are proposed to be responsible for the alteration of the insulin signaling molecules are post-transcriptional modifications. Various kinases are able to phosphorylate the substrate of the insulin receptor. This phosphorylation is responsible for the inhibition of this receptor [66]. TNF- α is a proinflammatory cytokine effector that is over-expressed in obese patients. It can activate JNKs and, therefore, mediate IR by inhibiting insulin signaling in the liver [67].
On the other hand, chronic high concentrations of glucose and leptin induce the secretion of IL-1β from the pancreatic islet, that can also possibly promote β-cell malfunction and death. This mechanism is also mediated by the JNK pathway. Therefore, the inhibition of JNK might protect the pancreatic β-cells from the outcomes that high levels of leptin and glucose have in diabetic patients.
Another reason that leads to insulin resistance is chronic oxidative stress. This mechanism is linked to JNKs that can mediate the consequences that stress has on insulin resistance. This mediation is due to the inhibition of the phosphorylation of insulin receptor substrate 1 (IRS-1). In addition, free fatty acids are able to activate, via JNKs, autophagy in pancreatic β-cells.
3.2. Adipokines
Studies have shown that adipokines (cytokines secreted by the adipocytes) are increased in NAFLD patients.
Yilmaz et al. investigated the patients with histological confirmed NAFLD regarding the relationship between the thickness of epicardial fat (EFT), adipokines related to epicardial fat, and coronary flow reserve (CFR). Fifty-four subjects with NAFLD (26 males and 28 females) and 56 control subjects (27 males and 29 females) were analyzed. NAFLD was evaluated by endoscopic ultrasound-guided biopsies. CFR and EFT were assessed by transthoracic cardiac ultrasonography. In addition, enzyme-linked immunosorbent assay (ELISA) was used in order to measure serum levels of vaspin and chemerin. The results proved that CFR was notably lower and EFT significantly higher in subjects with fatty liver disease than in controls. In addition, patients with NAFLD had serum levels of chemerin and vaspin significantly increased compared to controls [68].
Some examples of adipokines are leptin, adiponectin, resistin, and TNF-α. Because adipokines are involved in insulin resistance and inflammation, they might also be involved in NAFLD pathogenesis. TNF-α intercedes with insulin signaling and has also proinflammatory action. It has a major role in the apoptotic and proinflammatory responses to endotoxin [69]. Adiponectin is a protein that has completely opposite effects to TNF-α and also suppresses its secretion [70]. Regarding the lipidic metabolism, adiponectin has anti-lipogenic effects and reduces fat deposition. These effects are due to the inhibition of hepatic gluconeogenesis and also to the suppression of lipogenesis [71]. It has been shown that patients with NAFLD have hypoadiponectinemia, which is linked to CAD and diminished glucose tolerance in non-diabetic individuals [72][73].
3.3. Oxidative Stress
The factors that are proposed to be responsible for oxidative stress are hyperinsulinemia, bacterial overgrowth, hepatic iron, and lipid peroxidation [74].
Bacterial overgrowth might increase the hepatic oxidative stress by increasing the endogenous manufacturing of ethanol. Moreover, other mechanisms that may be involved in this process are the activation of inflammatory cytokines and macrophages [75].
It has been shown that patients that undergo chronic peritoneal dialysis only develop hepatic steatosis when insulin is infused to the peritoneal fluid dialysis [76][77]. This effect might be due to the ability of insulin to generate oxidative stress [78]. Another explanation is that insulin has the ability to up-regulate the lipogenic protein, sterol regulatory element-binding protein (SREBP) [79]. On the other hand, recent studies show that insulin might also be involved in apoptosis. This process might be due to insulin’s capacity to cause stress to endoplasmic reticulum, which leads to unfolding protein response [80].
Regarding iron’s prooxidant role, its contribution is still unclear. The only data that comes into the support for its oxidant role suggests that almost 30% of the NAFLD patients have elevated ferritin [81][82][83]. In addition, it has been reported that phlebotomy improves hepatic histology in patients with NAFLD [84]. Further investigations still need to be carried out regarding iron’s role in NAFLD.
3.4. Lipid Peroxidation and Apoptosis
The starting point of oxidative stress within the liver in patients with fatty liver disease are the free fatty acids (FFAs). Elevated FFAs act as ligands for peroxisome proliferator-activated receptor, which is in charge of the up-regulating of FFAs oxidation. This process take place in mitochondria, microsomes, and peroxisomes. FFAs oxidation results in hydrogen peroxide, superoxide, and lipid peroxides that generate oxidative stress and consequent lipid peroxidation [85].
Apoptosis is a mechanism that has multiple pathophysiologic causes and takes part in the development of liver injury and steatosis [74]. It affects hepatocytes and natural killer T cells. The causes of apoptosis in NAFLD are increased TNF, hyperinsulinemia, and oxidative stress. TNF serum levels are higher in patients with NAFLD and obesity. It is responsible for increased mitochondrial permeability, impaired mitochondrial respiration, and depleted mitochondrial cytochrome, all mechanisms leading to apoptosis [86][87].
As it was discussed before, hyperinsulinemia is another mechanism responsible for apoptosis. This effect might be due to insulin’s capacity to generate oxidative stress or up-regulate the lipogenic protein, SREBP [78][79]. On the other hand, hyperinsulinemia might also have direct effects on fibrogenesis by increasing the activity of connective tissue growth factor, mainly if hyperglycemia is present [88].
Regarding oxidative stress, malondialdyde (MDA) and hydroxynonenal (HNE) are two of the byproducts. They are able to attract neutrophils, to act as stimulants for hepatic stellate cells and to increase the number of receptors for cytokines and transforming growth factor-β (TGF-β) in macrophages [89]. Oxidative stress can also stimulate the discharge of TNF from the hepatic cells, lipocytes, and macrophages [89]. Hepatic stellate cells have the capacity to engulf the apoptotic bodies, a process that might stimulate their fibrogenic process [90]. Another mechanism that is responsible for hepatocyte deaths is ROS-induced fatty acid synthase (FAS) ligand [91][92].
In conclusion, there is a direct connection between NAFLD and CAD. Despite the fact that the pathophysiologic mechanisms behind this correlation still need further investigation, it is clear that a multidisciplinary approach of these patients must be taken into consideration.
4. Non-Alcoholic Fatty Liver Disease and Cardiac Arrhythmias
4.1. Atrial Fibrillation
In the OPERA (Oulu Project Elucidating Risk of Atherosclerosis) prospective study, 958 hypertensive subjects and control subjects of the same age and sex were arbitrarily selected from the national registries in 1990s, and the association between atrial fibrillation (AF) and NAFLD was determined. NAFLD was diagnosed by sonography and AF was tracked in the National Registers. The aim of this research was to evaluate the capacity of NAFLD to predict AF. The subjects included in this study were followed-up for 16.3 years. The authors concluded that NAFLD was an independent predictor of AF [93]. Another prospective study carried out by Giovanni et al. followed 400 patients with T2DM over 10 years. The subjects had no AF to start with. They also found out that NAFLD is related to a higher incidence of AF [94].
4.2. Ventricular Arrhythmias, Bundle Branch, and Atrioventricular Blocks
Recent data have suggested that type 2 diabetes patients with NAFLD have a higher chance of heart rate-corrected QT (QTc) interval prolongation. QT prolongation is a known risk factor for ventricular arrhythmias and sudden cardiac death.
Hung et al. carried out a cross-sectional study that included 31,116 participants. QTc interval was derived from Bazett’s formula and from electrocardiography. Ultrasonography was used to diagnose and classify NAFLD. NAFLD was staged as severe, moderate, mild, or none. The analyses found that mild, moderate, and severe NAFLD were associated with increased QTc interval compared to no NAFLD. The conclusions were that NAFLD was indeed associated with an increased risk of prolonged QTc interval among general population, independently of the presence of diabetes [95].
Another study carried out by Mantovani et al. has retrospectively evaluated 330 patients with T2DM and no end-stage renal disease, preexisting atrial fibrillation, or known liver diseases. The patients undergone 24-h Holter monitoring between 2013 and 2015. NAFLD was diagnosed by ultrasonography. Their study concluded that NAFLD was linked to a higher risk of prevalent ventricular arrhythmias in subjects with T2DM [96].
Matovani et al. carried out a retrospective study assessing whether there is any association connecting fatty liver disease and heart blocks. Seven hundred and fifty-one patients with T2DM were examined during 2007–2014. Atrioventricular blocks were assessed by electrocardiogram and NAFLD was diagnosed by ultrasonography. The study concluded that patients with fatty liver disease had a much higher risk of persistent heart block than those without NAFLD [97].
4.3. Mechanisms Behind Non-Alcoholic Fatty Liver Disease and Cardiac Arrhythmias
Currently, it is difficult to ignore the possibility that similar risk factors might be the cause behind cardiac arrhythmias and NAFLD. Obesity is generally associated with NAFLD and AF development [98][99]. On the other hand, both obesity and NAFLD are strongly associated with the accumulation of fat in the epicardium. Epicardial fat is linked to an increased mass of the left ventricle and diastolic dysfunction, the latter being associated with the promotion of AF [100]. Recent studies link NAFLD and diastolic dysfunction. The mechanisms behind this association might be extensive toxic results, mediated through adiponectin, insulin, and inflammation or indirect effects of hypertension or diabetes [101].
On the other hand, NAFLD is also associated with autonomic nervous system dysfunction and so is epicardial fat, dysfunction that is a risk factor for AF [102][103].
Regarding systemic inflammation, there are studies revealing that AF is as most a cause of systemic inflammation as it is a consequence [104], and, as it was discussed before, NAFLD is also associated with systemic inflammation. Moreover, hypoadiponectinemia, a usual finding in NAFLD is also related to AF [105]. This might explain how obesity and inflammation seem to play an important part in the relationship between NAFLD and AF. Taking all this information into consideration, we are still unable to differentiate if NAFLD is a cause of AF without the association of metabolic syndrome or if AF can itself contribute to the progression of NAFLD. Therefore, further studies are still needed in order to evaluate the real contribution of NAFLD to AF. Figure 1 presents the way that NAFLD and obesity increase the risk of developing hypertension.
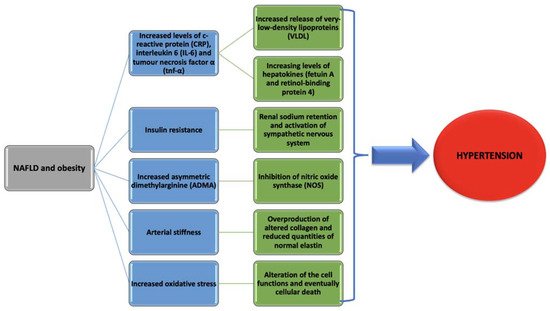
Figure 1. Pathogenic mechanisms behind hypertension development in non-alcoholic fatty liver disease (NAFLD) and obesity.
One study carried out by Lin YK et al. analyzed how adipocytes modulate the electrophysiology of atrial myocytes [14]. They used whole-cell patch clamp in order to record the action potentials and ionic currents in myocytes coming from rabbit left atrium. Myocites were incubated with and without adipocytes belonging to different sites of the body or adipocytes-conditioned supernatant for 2–4 h. Compared to control left atrium myocytes, left atrium myocytes incubated with adipocytes had longer action potentials durations. Left atrium myocytes incubated with adypocites from the epicardium had a more positive resting membrane potential than control left atrium myocytes. However, left atrium myocytes that were incubated with supernatant had longer action potential durations but similar resting membrane potential compared to control myocytes. Moreover, isoproterenol induced a higher incidence of provoked heart beats in left atrium myocytes that were incubated with adipocytes compared to the control group myocytes. All these taken into consideration, they concluded that adipocytes are able to cause arrhythmic heart beats in the myocytes of the left atrium. This might be possible due to adipocytes’ capacity to modulate electrophysiological characteristics and ion currents [106].
Moreover, pro-inflammatory cytokines and protrombotic factors in NAFLD are also linked to increased cardiac structural and arrhythmogenic complications [107].
On the other hand, NAFLD has been shown to be involved in the modifications of the myocardium functions and structure [108]. These modifications may induce fiber discontinuity and circuit re-entry, leading to electrophysiological disturbance [109].
In closing, it might be stated that there is indeed a powerful association between NAFLD and cardiac arrhythmias. Whether the common risk factors of these two entities are the cause or whether NAFLD is itself responsible for cardiac arrhythmias is still poorly investigated and further studies need to be carried on.
5. Non-Alcoholic Fatty Liver Disease and Altered Cardiac Structure
Multiple studies have reported the coexistence of NAFLD and altered cardiac structure. These modifications, regarding heart involve:
-
abnormalities in cardiac metabolism;
-
increased left ventricular mass;
-
increased interventricular septum thickness;
-
diastolic cardiac dysfunction;
-
left atrium enlargement or impaired left atrium deformation;
-
decreased right ventricular function;
-
aortic and mitral valves calcification;
-
congestive heart failure [110].
5.1. Abnormalities in Heart Metabolism
Perseghin et al. carried out a study assessing if subjects with fatty liver disease have also modifications regarding the left ventricle (LV). The modifications studied where those concerning the amounts of epicardial fat, the structure, energy metabolism, and function. Forty-two young, non-diabetic men, including 21 with NAFLD and 21 without NAFLD, were investigated. Cardiac MRI, cardiac 31phosphorus-magnetic resonance spectroscopy (31P-MRS) and hepatic 1Hydrogen-MRS (1H-MRS) were used as diagnostic tools. The updated Homeostasis Model Assessment (HOMA-2) computer model was used to determine insulin sensitivity. Systolic and diastolic functions, as well as left ventricular morphology, were not different among the two groups. On the other hand, the authors found out that intrapericardial and extrapericardial adipose tissue was increased in male subjects with fatty liver disease correlated to controls. The phosphocreatine (PCr)/adenosine triphosphate (ATP) ratios is an in vivo marker of myocardial energy metabolism. These ratios were reduced in men with NAFLD. Therefore, they concluded that although the morphological features and systolic and diastolic functions of the LV were normal, the energy metabolism was altered [111].
Despite the studies that show consistent evidence regarding altered heart metabolism, the pathogenic mechanism behind this is ambiguous. Ectopic liver fat accumulation is thought to be due to the increased free fatty acids flux, as it was showed before. Regarding the myocardium, it is known that its main source of energy in the fasting state are FFAs [112]. When the supply of FFAs outreaches the rate of their oxidative disposal, as happens in NAFLD, oxidative stress is induced, with the consequence of increased lipid intermediates and lipotoxicity. Lipotoxicity may be involved in the impairment of energy homeostasis and contractile dysfunction [113]. Other theories that might explain this perturbation, regarding the heart metabolism are the involvement of adipokines and inflammation. Because none of these hypotheses could be confirmed by this study, insulin resistance was thought to also be involved.
Concluding, although strong association between abnormalities in the heart metabolism and NAFLD exists, further studies providing information regarding the pathophysiology still need to be carried out.
5.2. Increased Left Ventricular Mass and Interventricular Septum Thickness
In a recent study, Hayrullah et al. assessed the grades of NAFLD and the cardiac functions and associated parameters [114]. Four hundred obese children participated in the study. Ultrasonography was used in order to diagnose NAFLD. Ninety-three children had NAFLD, 307 were in the non-NAFLD group, and these two subgroups were compared to 150 control subjects. In addition, pulsed and tissue Doppler echocardiography were used, and intima-media and epicardial adipose tissue were measured. Fatty liver disease subgroups had increased end-systolic thickness of the interventricular septum and larger left ventricular mass. Moreover, carotid artery intima-media and epicardial adipose tissue thickness were higher in obese children.
One pathologic mechanism that might explain these modifications is the eccentric remodeling of the LV as an effect of adaptation to volume overload. Moreover, central fat distribution is associated to higher cardiac output [115].
On the other hand, it is established that insulin resistance increases the chances for LV dysfunction development. Obese patients have increased levels of insulin and altered insulin sensibility, which is also related to NAFLD, as it was discussed before. Elevated insulin levels stimulate the growth of myocytes and interstitial fibrosis, by sodium retention and activation of sympathetic nervous system [116].
A meta-analysis carried out by Borges et al. noted an important relationship between NAFLD and higher left atrium diameter and ratio between left atrial volume and body surface area [117]. The left atrium is responsible for the filling of the left ventricle, and, on the other hand, the left ventricle function influences the left atrium’s function throughout the cardiac cycle [118]. As a consequence of eccentric remodeling of the LV, LV’s filling pressure increases and leads to left atrium dilatation. Therefore, left atrium remodeling occurs and atrial compliance and contractile functions decrease [119].
5.3. Decreased Right Ventricular Function
Bekler et al. studied systolic and diastolic function of the right ventricle and its association with NAFLD [120]. Thirty-two patients had NAFLD diagnosed by ultrasonography, and 22 represented the control group, i.e., had no NAFLD. The function of the right ventricle was assessed using conventional and tissue Doppler echocardiography. Right ventricle global function was evaluated by myocardial performance index (MPI). They concluded that right ventricular diastolic dysfunction correlated with fatty liver disease and the level of fatty changes.
Regarding this association, there are speculations assuming that the promotion of right ventricular diastolic function is due to the excess of lipid accumulation in hepatocytes, that leads to lipid deposition in cardiac myocytes.
Aortic and mitral valves calcification: Several studies have demonstrated that NAFLD associates with calcium deposits of the aorta and mitral valves [121].
Aortic valve sclerosis is associated with a higher incidence of cardiovascular disease mortality. This statement is valid for both diabetic and non-diabetic patients. In addition, the calcium deposits in the mitral valve are associated with adverse cardiovascular disease outcomes [122]. One mechanism that may explain the link between NAFLD and the calcification of aortic and mitral valves is chronic inflammation. It was discussed in the previous paragraphs how the liver produces inflammatory markers. These are believed to contribute to the acceleration of atherosclerosis, as it is demonstrated in histopathological examinations, which show that valve sclerosis is an inflammatory condition mixed with classic atherosclerotic lesions [123][124].
In addition, insulin resistance might be responsible for the association between hepatic steatosis and aortic valve sclerosis. One small cross-sectional study concluded that increased steatosis of the liver is associated with lower insulin clearance, which contributes to insulin resistance in non-diabetic subjects [125]. However, another study carried out by Marcello et al. [126] provided evidence that clinical diabetes mellitus did not substantially affected the results.
5.4. Diastolic Cardiac Dysfunction and Risk of Congestive Heart Failure
Explanations behind diastolic cardiac dysfunction and NAFLD are systemic inflammation, insulin resistance and increased prevalence of metabolic disorders.
Systemic inflammation is mediated by NAFLD, as it was discussed before. Cytokines are able to modify the structural substrate and electrophysiology of the myocardium. Moreover, this pro-inflammatory condition is associated with inflammation in the coronary microvascular endothelium. This is able to increase the LV diastolic stiffness and lead to cardiac failure [127].
Insulin resistance, a common comorbidity among patients with NAFLD and obesity, alters diastolic function. This is possible due to insulin’s ability to regulate the expression of myosin gene [128].
Metabolic disorders, such as hypertension, are risk factors for diastolic cardiac dysfunction and are frequently associated with NAFLD.
Taking all the information above into consideration, it is likely to assume that fatty liver disease is possibly linked to a higher chance of developing congestive heart failure. Several researches that used elevated aminotransferases levels or serum gamma-glutamyl transferase (GGT) as markers of NAFLD have proven that this disorder is strongly connected to heart failure [129][130].
Regarding GGT, the mechanisms that may speculatively be involved in this association are the presence of GGT in atherosclerotic plaques and that elevated GGT levels might be a marker of increased oxidative stress [131].
6. Non-Alcoholic Fatty Liver Disease and Stroke
Multiple studies have been evaluating over the years the correlation between NAFLD and the presence of stroke.
Hamaguchi et al. have prospectively analyzed 1637 apparently healthy patients. NAFLD was determined by ultrasound. After five years of assessments, a self-administered questionnaire was used to evaluate the incidence of cardiovascular disease. Between the 1221 subjects in attendance for results analysis, the occurrence of cardiovascular disease was elevated in the group composed of the subjects that had NAFLD to start with than the group without NAFLD [132].
Another prospective study carried out by Abdeldyem et al. evaluated the anticipating value that NAFLD has on stroke gravity and prognostic. Two hundred subjects that suffered an acute ischemic stroke were studied. Regarding NAFLD, the diagnosis was based on the elevated levels of serum aminotransferases and the lack of any other causes that are able to increase them. They concluded that NAFLD was associated with acute ischemic stroke in 42.5% of the subjects. Moreover, NAFLD might also be related to even worse gravity and prognostic of stroke [133].
Regarding the pathophysiology of the correlation between NAFLD and stroke, evidence suggests a relationship between ischemic stroke and biological markers of NAFLD [134]. These biological markers are represented by AST, ALT, and γ-glutamyltransferase (GGT).
A study by Bots et al. analyzed how GGT, as a marker of alcohol consumption associated with hemorrhagic, ischemic, fatal and non-fatal stroke. Three European cohort studies were included, taking part in EUROSTROKE. They concluded that an increased GGT level was linked to an increased chance of developing hemorrhagic stroke [135]. As it was previously discussed, GGT is an enzyme that is presented in the atherosclerotic lesions. GGT locates in the CD68 macrophage-derived foam cells [136]. In addition, other evidences show that GGT is identified in the circulating platelets and granulocytes [137][138]. On the other hand, GGT seems to be adsorbed into circulating LDL. The main role of GGT is the degradation of glutathione, which is an important antioxidant. In selected conditions, GGT plays a prooxidant role. Moreover, it has been demonstrated that GGT-mediated cleavage of glutamate-cysteine-glycine (GSH) can affect the reduction of ferric iron to ferrous iron, this process being able to produce reactive oxygen species [139]. Oxidative reactions are important determinants of the plaque development and instability [140]. They are able to regulate matrix metalloproteinases, which are key factors in determining the plaque stability.
After all these presented before, it can be stated that GGT, which is an important constituent of the atherogenic plaques, might be a major pro-oxidant component and also a marker of stroke.
7. Non-Alcoholic Fatty Liver Disease and Thromboembolic Events
Recent studies proposed that NAFLD patients have an increased chance of developing thromboembolic events regardless of the presence of diabetes, hyperlipidemia, and obesity that are usually associated to NAFLD. Although there are few statistical analysis concerning the associations between fatty liver disease and thromboembolism, recent studies have reported a higher risk of portal venous and systemic thrombosis in forms of advanced NAFLD disease [141].
A case-control study carried by Di Minno et al. documented idiopathic venous thromboembolism (VTE) in 138 patients, with 276 subjects being used as controls. All of them underwent clinical/laboratory/ultrasound evaluation for the presence of metabolic syndrome and NAFLD. One hundred and twelve out of 138 cases and 84/276 controls were diagnosed with VTE. Therefore, they concluded that NAFLD was independently associated with idiopathic VTE [142].
Stine et al. used a cross-sectional research for the evaluation of autonomous association between NASH cirrhosis and portal vein thrombosis (PVT) in patients who experienced hepatic transplantation. T\hirty-three thousand three hundred and sixty-eight patients were included in the study. Of these, 6.3% had PVT. Among them, 12.0% had NASH. When these subjects were compared to another group with different causes of cirrhosis, it was found that the subjects that underwent transplantation had a higher prevalence of PVT. Ten and one tenth percent were having PVT at the time of transplantation, compared to 6.0% without NASH [143].
Metabolic syndrome: As it was discussed before, metabolic syndrome or X syndrome is, as well, a cause, as it is a consequence of NAFLD. X syndrome is a condition that identifies with hypercoagulation, platelet activation and endothelial dysfunction, all those mechanisms being responsible for the occurrence of atherosclerosis. Moreover, metabolic syndrome is also associated with the reduction of anticoagulant proteins and the inhibition of fibrinolysis [144].
Insulin resistance: Insulin resistance in NAFLD is responsible for the increased levels of triglycerides, which is, in turn, responsible for promoting hypercoagulation by elevating the coagulation factors [145].
Moreover, impaired fasting glucose and diabetes are associated with a higher risk of developing cardiovascular diseases and arterial thrombosis [146]. The mechanisms behind this association are thought to be increased thrombin generation, dysfunctional activation of the platelets, calcium release, and inositol phospholipid turnover [147][148].
In addition, hyperglycemia is also responsible for altered coagulation cascade by inducing oxidative stress, decreasing plasmatic heparin sulphate and by the non-enzymatic glycation of proteins [149].
Another factor that contributes to the prothrombotic state is NO deficiency, this monoxide being responsible for the inhibition of platelets’ aggregation [150]. As it was discussed before, insulin resistance is responsible for limiting the NO production.
TNF-α, which is increased in NAFLD, impairs the synthesis of endothelial NOS (eNOS). eNOS contributes to a lower amount of NO compared to inducible NOS (iNOS). Several cell types express iNOS, which generates NO as a result of inflammatory state. In this regard, during the inflammatory state, the suppression of iNOS was associated with the diminution of coagulation [151].
Gut microbiota: Studies have shown that a diet rich in fats in mice that develop NAFLD altered the manufacturing of metabolites dependent on intestinal flora, including trimethylamine (TMA) [152]. The relationship within TMA and NAFLD has also been verified in humans [153]. TMA enhances the formation of foam cells from macrophages and platelet activation [154].
Plasminogen activator inhibitor-1 (PAI-1): PAI-1 suppresses tissue plasminogen activator (tPA), therefore reducing the activity of fibrinolysis. NAFLD is linked to higher concentrations of PAI-1, therefore coming into the support of another cause of prothrombotic state [155].
Thromboinflammation, thrombocytes, and activated endothelium: Thrombosis and inflammation are firmly joint. Higher C-reactive protein levels are responsible for the promotion of the interaction between the monocytes and the endothelium and for the activity of PAI-1 and formation of the tissue factor. As a result, the natural anticoagulant systems are down-regulated [156].
NAFLD is associated with a high inflammatory state. This proinflammatory condition is also marked by thrombocytes activation and endothelial impaired function. Endothelial cells produce von Willebrand factor (vWF), factor VIII, and fibrinogen, that are, both, inflammatory and clotting elements.
P-selectin glycoprotein ligand-1 and P-selectin mediate the contact between platelets and endothelium [157]. In addition, adhesion molecules, such as intercellular adhesion molecule one-1 (ICAM-1), are involved in this process, and it has been shown that plasma levels of ICAM-1 are increased in NAFLD [155].
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84.
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver. Dis. 2018, 22, 11–21.
- Zhu, J.Z.; Hollis-Hansen, K.; Wan, X.Y.; Fei, S.J.; Pang, X.L.; Meng, F.D.; Yu, C.H.; Li, Y.M. Clinical Guidelines of Non-Alcoholic Fatty Liver Disease: A Systematic Review. World J. Gastroenterol. 2016, 22, 8226–8233.
- Saverymuttu, S.H.; Joseph, A.E.; Maxwell, J.D. Ultrasound Scanning in the Detection of Hepatic Fibrosis and Steatosis. Br. Med. J. Clin. Res. 1986, 292, 13–15.
- Thomas, E.L.; Hamilton, G.; Patel, N.; O’Dwyer, R.; Dore, C.J.; Goldin, R.D.; Bell, J.D.; Taylor-Robinson, S.D. Hepatic Triglyceride Content and Its Relation to Body Adiposity: A Magnetic Resonance Imaging and Proton Magnetic Resonance Spectroscopy Study. Gut 2005, 54, 122–127.
- Tang, A.; Desai, A.; Hamilton, G.; Wolfson, T.; Gamst, A.; Lam, J.; Clark, L.; Hooker, J.; Chavez, T.; Ang, B.D.; et al. Accuracy of MR Imaging Estimated Proton Density Fat Fraction for Classification of Dichotomized Histologic Steatosis Grades in Nonalcoholic Fatty Liver Disease. Radiology 2015, 274, 416–425.
- Yoshio, S.; Atsushi, N.; Yoshito, I. Limitations of Liver Biopsy and Non-Invasive Diagnostic Tests for the Diagnosis of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. World J. Gastroenterol. 2014, 20, 475–485.
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic Steatohepatitis: A Proposal for Grading and Staging the Histological Lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474.
- Neuman, M.G.; Malnick, S.; Maor, Y.; Nanau, R.M.; Melzer, E.; Ferenci, P.; Seitz, H.K.; Mueller, S.; Mell, H.; Samuel, D.; et al. Alcoholic Liver Disease: Clinical and Translational Research. Exp. Mol. Pathol. 2015, 99, 596–610.
- Park, S.H.; Jeon, W.K.; Kim, S.H.; Kim, H.J.; Park, D.I.; Cho, Y.K.; Sung, I.K.; Sohn, C.I.; Keum, D.K.; Kim, B.I. Prevalence and Risk Factors of Non-Alcoholic Fatty Liver Disease among Korean Adults. J. Gastroenterol. Hepatol. 2006, 21, 138–143.
- Anstee, Q.M.; Targher, G.; Day, C.P. Nat Rev Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Gastroenterol. Hepatol. 2013, 10, 330–344.
- Yan-Ci, Z.; Guo-Jun, Z.; Ze, C.; Zhi-Gang, S.; Jingjing, C.; Hongliang, L. Nonalcoholic Fatty Liver Disease An Emerging Driver of Hypertension. Hypertension 2020, 75, 275–284.
- Catena, C.; Bernardi, S.; Sabato, N.; Grillo, A.; Ermani, M.; Sechi, L.A.; Fabris, B.; Carretta, R.; Fallo, F. Ambulatory Arterial Stiffness Indices and Nonalcoholic Fatty Liver Disease in Essential Hypertension. Nutr. Metab. Cardiovasc. Dis. 2012, 23, 389–393.
- Lin, Y.C.; Lo, H.M.; Chen, J.D. Sonographic Fatty Liver, Overweight and Ischemic Heart Disease. World J. Gastroenterol. 2005, 11, 4838.
- Schindhelm, R.K.; Dekker, J.M.; Nijpels, G.; Bouter, L.M.; Stehouwer, C.D.A.; Heine, R.J.; Diamant, M. Alanine Aminotransferase Predicts Coronary Heart Disease Events: A 10-Year Follow-up of the Hoorn Study. Atherosclerosis 2007, 191, 391–396.
- Sinn, D.H.; Gwak, G.Y.; Park, H.N.; Kim, J.E.; Min, Y.W.; Kim, K.M.; Kim, Y.J.; Choi, M.S.; Lee, J.H.; Koh, K.C.; et al. Ultrasonographically Detected Non-Alcoholic Fatty Liver Disease Is an Independent Predictor for Identifying Patients with Insulin Resistance in Non-Obese, Non-Diabetic Middle-Aged Asian Adults. Am. J. Gastroenterol. 2012, 107, 561–567.
- Musso, G.; Gambino, R.; Bo, S.; Uberti, S.; Biroli, G.; Pagano, G.; Cassader, M. Should Nonalcoholic Fatty Liver Disease Be Included in the Definition of Metabolic Syndrome? A Cross-Sectional Comparison with Adult Treatment Panel III Criteria in Nonobese Nondiabetic Subjects. Diabetes Care 2008, 31, 562–568.
- Haukeland, J.W.; Damås, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic Inflammation in Nonalcoholic Fatty Liver Disease Is Characterized by Elevated Levels of CCL2. J. Hepatol. 2006, 44, 1167–1174.
- Stumpf, C.; Auer, C.; Yilmaz, A.; Lewczuk, P.; Klinghammer, L.; Schneider, M.; Daniel, W.G.; Schmieder, R.E.; Garlichs, C.D. Serum Levels of the Th1 Chemoattractant Interferon-γ-inducible Protein (IP) 10 Are Elevated in Patients with Essential Hypertension. Hypertens. Res. 2011, 34, 484–488.
- Bai, L.; Li, H.I. Immune Regulatory Networks in Hepatic Lipid Metabolism. J. Mol. Med. Berl. 2019, 97, 593–604.
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking Nonalcoholic Fatty Liver Disease and Insulin Resistance. Nat. Rev. Endocrinol. 2017, 13, 509.
- Nunes, K.P.; de Oliveira, A.A.; Mowry, F.E.; Biancardi, V.C. Targeting Toll-like Receptor 4 Signalling Pathways: Can Therapeutics Pay the Toll for Hypertension? Br. J. Pharmacol. 2019, 176, 1864–1879.
- Sinn, D.H.; Kang, D.; Jang, H.R.; Gu, S.; Cho, S.J.; Paik, S.W.; Ryu, S.; Chang, Y.; Lazo, M.; Guallar, E.; et al. Development of Chronic Kidney Disease in Patients with Non-Alcoholic Fatty Liver Disease: A Cohort Study. J. Hepatol. 2017, 67, 1274–1280.
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The Impact of Insulin Resistance on the Kidney and Vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737.
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393.
- Landsberg, L.; Young, J.B. Insulin Mediated Glucose Metabolism in the Relationship between Dietary Intake and Sympathetic Nervous System Activity. Int. J. Obes. 1985, 9, 63–68.
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed Hepatic Bile Acid Signalling despite Elevated Production of Primary and Secondary Bile Acids in NAFLD. Gut 2018, 67, 1881–1891.
- Kasumov, T.; Edmison, J.M.; Dasarathy, S.; Bennett, C.; Lopez, R.; Kalhan, S.C. Plasma levels of asymmetric dimethylarginine (ADMA) in patients with biopsy-proven non-alcoholic fatty liver disease. Metabolism 2011, 60, 776–781.
- Serg, M.; Kampus, P.; Kals, J.; Zagura, M.; Muda, P.; Tuomainen, T.P.; Zilmer, K.; Salum, E.; Zilmer, M.; Eha, J. Association between Asymmetric Dimethylarginine and Indices of Vascular Function in Patients with Essential Hypertension. Blood Press 2011, 20, 111–116.
- Sydow, K.; Mondon, C.E.; Cooke, J.P. Insulin Resistance: Potential Role of the Endogenous Nitric Oxide Synthase Inhibitor ADMA. Vasc. Med. 2005, 10 (Suppl. 1), S35–S43.
- Abbasi, F.; Asagmi, T.; Cooke, J.P.; Lamendola, C.; McLaughlin, T.; Reaven, G.M.; Stuehlinger, M.; Tsao, P.S. Plasma Concentrations of Asymmetric Dimethylarginine Are Increased in Patients with Type 2 Diabetes Mellitus. Am. J. Cardiol. 2001, 88, 1201–1203.
- Villela-Nogueira, C.A.; Leite, N.C.; Cardoso, C.R.L.; Salles, G.F. NAFLD and Increased Aortic Stiffness: Parallel or Common Physiopathological Mechanisms? World J. Gastroenterol. 2014, 14, 8377–8392.
- Johnson, C.P.; Baugh, R.; Wilson, C.A.; Burns, J. Age Related Changes in the Tunica Media of the Vertebral Artery: Implications for the Assessment of Vessels Injured by Trauma. J. Clin. Pathol. 2001, 54, 139–145.
- Nickenig, G.; Roling, J.; Strehlow, K.; Schnabel, P.; Bohm, M. Insulin Induces Upregulation of Vascular AT1 Receptor Gene Expression by Posttranscriptional Mechanisms. Circulation 1998, 8, 2453–2460.
- Jesmin, S.; Sakuma, I.; Salah-Eldin, A.; Nonomura, K.; Hattori, Y.; Kitabatake, A. Diminished Penile Expression of Vascular Endothelial Growth Factor and Its Receptors at the Insulin-Resistant Stage of a Type II Diabetic Rat Model: A Possible Cause for Erectile Dysfunction in Diabetes. J. Mol. Endocrinol. 2003, 31, 401–418.
- Rizzoni, D.; Porteri, E.; Guelfi, D.; Muiesan, M.L.; Valentini, U.; Cimino, A.; Girelli, A.; Rodella, L.; Bianchi, R.; Sleiman, I.; et al. Structural Alterations in Subcutaneous Small Arteries of Normotensive and Hypertensive Patients with Non-Insulin-Dependent Diabetes Mellitus. Circulation 2001, 103, 1238–1244.
- Vlachopoulos, C.; Manesis, E.; Baou, K.; Papatheodoridis, G.; Koskinas, J.; Tiniakos, D.; Aznaouridis, K.; Archimandritis, A.; Stefanadis, C. Increased Arterial Stiffness and Impaired Endothelial Function in Nonalcoholic Fatty Liver Disease: A Pilot Study. Am. J. Hypertens. 2010, 23, 1183–1189.
- Kim, B.J.; Kim, N.H.; Kim, B.S.; Kang, J.H. The Association between Nonalcoholic Fatty Liver Disease, Metabolic Syndrome and Arterial Stiffness in Nondiabetic, Nonhypertensive Individuals. Cardiology 2012, 123, 54–61.
- Huang, Y.; Bi, Y.; Xu, M.; Ma, Z.; Xu, Y.; Wang, T.; Li, M.; Liu, Y.; Lu, J.; Chen, Y.; et al. Nonalcoholic Fatty Liver Disease Is Associated with Atherosclerosis in Middle-Aged and Elderly Chinese. Arter. Thromb. Vasc. Biol. 2012, 32, 2321–2326.
- Lee, Y.J.; Shim, J.Y.; Moon, B.S.; Shin, Y.H.; Jung, D.H.; Lee, J.H.; Lee, H.R. The Relationship between Arterial Stiffness and Nonalcoholic Fatty Liver Disease. Dig. Sci. 2012, 57, 196–203.
- Ozturk, A.K.; Uygun, A.A.; Guler, B.A.K.; Demirci, A.H.; Ozdemir, C.C.; Cakir, B.M.; Sakin, A.Y.S.; Turker, D.T.; Sari, E.S.; Demirbas, B.S.; et al. Nonalcoholic Fatty Liver Disease Is an Independent Risk Factor for Atherosclerosis in Young Adult Men. Atherosclerosis 2015, 240, 380–386.
- Jain, S.; Khera, R.; Corrales-Medina, V.F.; Townsend, R.R.; Chirinos, J.A. Inflammation and Arterial Stiffness in Humans. Atherosclerosis 2014, 237, 381–390.
- Farzanegi, P.; Amir, D.; Ebrahimpoor, Z.; Mahdieh, A.; Azarbayjani, M.A. Mechanisms of Beneficial Effects of Exercise Training on Non-Alcoholic Fatty Liver Disease (NAFLD): Roles of Oxidative Stress and Inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003.
- Colagar, A.H.; Marzony, E.T. Ascorbic Acid in Human Seminal Plasma: Determination and Its Relationship to Sperm Quality. J. Clin. Biochem. Nutr. 2009, 45, 144–149.
- Yu, Y.; Cai, J.; She, Z.; Li, H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv. Sci. Weinh. 2019, 6, 1801585.
- Distrutti, E.; Mencarelli, A.; Santucci, L.; Renga, B.; Orlandi, S.; Donini, A.; Shah, V.; Fiorucci, S. The Methionine Connection: Homocysteine and Hydrogen Sulfide Exert Opposite Effects on Hepatic Microcirculation in Rats. Hepatology 2008, 47, 659–667.
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative Stress: New Insights on the Association of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336.
- Wang, Y.; Chen, S.; Yao, T.; Li, D.; Wang, Y.; Li, Y.; Wu, S.; Cai, J. Homocysteine; as a Risk Factor for Hypertension: A 2-Year Follow-up Study. PLoS ONE 2014, 9, e108223.
- Björkholm, B.; Bok, C.M.; Lundin, A.; Rafter, J.; Hibberd, M.L.; Pettersson, S. Intestinal Microbiota Regulate Xenobiotic Metabolism in the Liver. PLoS ONE 2009, 4, e6958.
- Safari, Z.; Gérard, P. The Links between the Gut Microbiome and Non Alcoholic Fatty Liver Disease (NAFLD). Cell. Mol. Life Sci. 2019, 76, 1541–1558.
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond Gut Feelings; How the Gut Microbiota Regulates Blood Pressure. Nat. Rev. Cardiol. 2018, 15, 20–32.
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911.
- Fan, W.; Qu, X.; Li, J.; Wang, X.; Bai, Y.; Cao, Q.; Ma, L.; Zhou, X.; Zhu, A.W.; Liu, B.W.; et al. Associations between Polymorphisms of the ADIPOQ Gene and Hypertension Risk: A Systematic and Meta-Analysis. Sci. Rep. 2017, 7, 41683.
- Zhu, W.; Cheng, K.K.; Vanhoutte, P.M.; Lam, K.S.; Xu, A. Vascular Effects of Adiponectin: Molecular Mechanisms and Potential Therapeutic Intervention. Clin. Sci. 2008, 114, 361–374.
- Musso, G.; Saba, F.; Cassader, M.; Paschetta, E.; De Michieli, F.; Pinach, S.; Framarin, L.; Berrutti, M.; Leone, N.; Parente, R.; et al. Angiotensin II Type 1 Receptor Rs5186 Gene Variant Predicts Incident NAFLD and Associated Hypertension: Role of Dietary Fat-Induced pro-Inflammatory Cell Activation. Am. J. Gastroenterol. 2019, 114, 607–619.
- Srivastava, R.A.K. Life-Style-Induced Metabolic Derangement and Epigenetic Changes Promote Diabetes and Oxidative Stress Leading to NASH and Atherosclerosis Severity. J. Diabetes Metab. Disord. 2018, 17, 381–391.
- Costantino, S.; Mohammed, S.A.; Ambrosini, S.; Paneni, F. Epigenetic Processing in Cardiometabolic Disease. Atherosclerosis 2019, 281, 150–158.
- Bungau, S.; Behl, T.; Tit, D.M.; Banica, F.; Bratu, O.G.; Diaconu, C.C.; Nistor-Cseppento, C.D.; Bustea, C.; Aron, R.A.C.; Vesa, C.M. Interactions between Leptin and Insulin Resistance in Patients with Prediabetes, with and without NAFLD. Exp. Ther. Med. 2020, 20, 197.
- Gheorghe, G.; Bungau, S.; Ceobanu, G.; Ilie, M.; Bacalbasa, N.; Bratu, O.G.; Vesa, C.M.; Gaman, M.A.; Diaconu, C.C. The Non-Invasive Assessment of Hepatic Fibrosis. J. Formos. Med. Assoc. 2021, 120, 794–803.
- Ampuero, J.; Gallego-Durán, R.; Romero-Gómez, M. Association of NAFLD with Subclinical Atherosclerosis and Coronary-Artery Disease: Meta-Analysis. J. Rev. Esp. Enferm. Dig. 2015, 107, 10–16.
- Yoosoo, C.; Seungho, R.; Ki-Chul, S.; Yong, K.C.; Eunju, S.; Han-Na, K.; Hyun-Suk, J.; Kyung, E.Y.; Jiin, A.; Hocheol, S.; et al. Alcoholic and Non-Alcoholic Fatty Liver Disease and Associations with Coronary Artery Calcification (CAC): Evidence from the Kangbuk Samsung Health Study. Gut 2019, 68, 1667–1675.
- Treeprasertsuk, S.; Lopez-Jimenez, F.; Lindor, K.D. Nonalcoholic Fatty Liver Disease and the Coronary Artery Disease. Dig. Sci. 2011, 56, 35–45.
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Järvinen Hannele, Y. Increased Liver Fat, Impaired Insulin Clearance, and Hepatic and Adipose Tissue Insulin Resistance in Type 2 Diabetes. Gastroenterology 2008, 135, 122–130.
- Tarantino, G.; Caputi, A. JNKs, Insulin Resistance and Inflammation: A Possible Link between NAFLD and Coronary Artery Disease. World J. Gastroenterol. 2011, 17, 3785–3794.
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of b Cell Mass in Response to High-Fat Diet. Cell. Metab. 2008, 8, 325–332.
- Weickert, M.O. Signalling Mechanisms Linking Hepatic Glucose and Lipid Metabolism. Pfeiffer. AF Diabetol. 2006, 49, 1732–1741.
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and Tumor Necrosis Factor-Alpha Mediate Free Fatty Acid-Induced Insulin Resistance in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 35361–35371.
- Yilmaz, Y.; Kurt, R.; Gurdal, A.; Alahdaba, Y.O.; Yonal, O.; Senates, E.; Polat, N.; Erend, F.; Imeryuz, N.; Oflaz, H. Circulating Vaspin Levels and Epicardial Adipose Tissue Thickness Are Associated with Impaired Coronary Flow Reserve in Patients with Nonalcoholic Fatty Liver Disease. Atherosclerosis 2011, 217, 125–129.
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez -Escalante, J.C.; Pons-Romero, F. Gene Expression of Tumor Necrosis Factor and TNF-Receptors, P55 and P75, in Nonalcoholic Steatohepatitis Patients. Hepatology 2001, 34, 1158–1163.
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-Induced Insulin Resistance in Mice Lacking Adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737.
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.L.; Cooper, G.J.S. The Fat-Derived Hormone Adiponectin Alleviates Alcoholic and Nonalcoholic Fatty Liver Diseases in Mice. J. Clin. Investig. 2003, 112, 91–100.
- McKimmie, R.L.; Daniel, K.R.; Carr, J.J.; Bowden, D.W.; Freedman, B.I.; Register, T.C.; Hsu, F.C.; Lohman, K.K.; Weinberg, R.B.; Wagenknecht, L.E. Hepatic Steatosis and Subclinical Cardiovascular Disease in a Cohort Enriched for Type 2 Diabetes: The Diabetes Heart Study. Am. J. Gastroenterol. 2008, 103, 3029–3035.
- Otsuka, F.; Sugiyama, S.; Kojima, S.; Maruyoshi, H.; Funahashi, T.; Sakamoto, T.; Yoshimura, K.; Kimura, K.; Umemura, S.; Ogawa, H. Hypoadiponectinemia Is Associated with Impaired Glucose Tolerance and Coronary Artery Disease in Non-Diabetic Men. Circ. J. 2007, 71, 1703–1709.
- Edmison, J.; McCullough, A.J. Pathogenesis of Non-Alcoholic Steatohepatitis: Human Data. Clin. Liver Dis. 2007, 11, 75–104.
- Solga, S.F.; Diehl, A.M. Nonalcoholic Fatty Liver Disease: Lumen-Liver Interactions and Possible Role for Probiotics. J. Hepatol. 2003, 38, 681–687.
- Wanless, I.R.; Bargman, J.M.; Oreopoullos, D.G.; Vas, S.I. Subcapsular Steatonecrosis in Response to Peritoneal Insulin Deliver: A Clue to the Pathogenesis of Steatonecrosis in Obesity. Mod. Pathol. 1989, 2, 69–74.
- Khalili, K.; Lan, F.P.; Hanbidge, A.E.; Muradali, D.; Oreopoulos, D.G.; Wanless, I.R. Hepatic Subcapsular Steatosis in Response to Intraperitoneal Insulin Delivery: CT Findings and Prevalence. AJR Am. J. Roentgenol. 2003, 180, 1601–1604.
- Goldstein, B.J.; Kalyankar, M.; Wu, X. Insulin Action Is Facilitated by Insulin-Stimulated Reactive Oxygen Species with Multiple Potential Signaling Targets. Diabetes 2005, 54, 311–321.
- Li, X.L.; Man, K.; Ng, K.T.; Lee, T.K.; Lo, C.M.; Fan, S.T. Insulin in UW Solution Exacerbates Hepatic Ischemia/Reperfusion Injury by Energy Depletion through the IRS-2/SREBP-1c Pathway. Liver Transpl. 2004, 10, 1172–1182.
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461.
- Fargion, S.; Mattioli, M.; Fracanzani, A.L.; Sampietro, M.; Tavazzi, D.; Fociani, P.; Taioli, E.; Valenti, L.; Fiorelli, G. Hyperferritinemia, Iron Overload, and Multiple Metabolic Alterations Identify Patients at Risk for Nonalcoholic Steatohepatitis. Am. J. Gastroenterol. 2001, 96, 2448–2455.
- Fernandez Real, J.M.; Casamitjana-Abella, R.; Ricart-Engel, W.; Arroyo, E.; Balanca, R.; Casamitjana-Abella, R.; Cabrero, D.; Fernandez-Castaner, M.; Soler, J. Serum Ferritin as a Component of the Insulin Resistance Syndrome. Diabetes Care 1998, 21, 62–68.
- Mendler, M.-H.; Turlin, B.; Moirand, R.; Jouanolle, A.-M.; Sapey, T.; Guyader, D.; le Gall, J.-Y.; Brissot, P.; David, V.; Deugnier, Y. Insulin Resistance-Associated Hepatic Iron Overload. Gastroenterology 1999, 117, 1155–1163.
- Riquelme, A.; Soza, A.; Nazal, L.; Martinez, G.; Kolbach, M.; Patillo, A.; Arellano, M.; Duarte, I.; Martinez, J.; Molgo, M.; et al. Histological Resolution of Steatohepatitis after Iron Depletion. Dig. Sci. 2004, 49, 1012–1015.
- Clarke, S.D. Nonalcoholic Steatosis and Steatohepatitis. I. Molecular Mechanism for Polyunsaturated Fatty Acid Regulation of Gene Transcription. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G865–G869.
- Tafani, M.; Schneider, T.G.; Pastorino, J.G.; Farber, J.L. Cytochrome Dependent Activation of Caspase 3 by Tumor Necrosis Factor Requires Induction of the Mitochondrial Permeability Transition. Am. J. Pathol. 2000, 156, 2111–2121.
- Pastorini, J.G.; Simbula, G.; Yamamoto, K.; Glascott, P.A., Jr.; Rothman, R.J.; Farber, J.L. Cytotoxicity of TNF Depends on Induction of the Mitochondrial Permeability Transition. J. Biochem. 1996, 271, 29792–29798.
- Paradis, V.; Perle, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High Glucose and Hyperinsulinemia Stimulate Connective Tissue Growth Factor Expression: A Potential Mechanism Involved in Progression to Fibrosis in Nonalcoholic Steatohepatitis. Hepatology 2001, 74, 738–744.
- Poli, G. Pathogenesis of Liver Fibrosis: The Role of Oxidative Stress. Mol Asp. Med. 2000, 21, 49–98.
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic Body Engulfment by a Human Stellate Cell Line Is Profibrogenic. Lab. Investig. 2003, 83, 655–663.
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte Apoptosis and FAS Expression Are Prominent Features of Human Nonalcoholic Steatohepatitis. Gastroenterology 2003, 125, 437–443.
- Li, Z.; Oben, J.A.; Yang, S.; Lin, H.; Stafford, E.A.; Soloski, M.J.; Thomas, S.A.; Diehl, A.M. Norepinephrine Regulates Hepatic Innate Immune System in Leptin Deficient Mice with Nonalcoholic Steatohepatitis. Hepatology 2004, 40, 434–441.
- Käräjämäki, A.J.; Olli-Pekka, P.; Markku, S.; Kesäniemi, Y.A.; Heikki, H.; Ukkola, O. Non-Alcoholic Fatty Liver Disease as a Predictor of Atrial Fibrillation in Middle-Aged Population (OPERA Study). PLoS ONE 2015, 10, e0142937.
- Targher, G.; Valbuso, F.; Bonapace, S.; Bertolini, L.; Zenari, L.; Rodella, S.; Zoppini, G.; Mantovani, W.; Barbieri, E.; Byrne, C.D. Non-Alcoholic Fatty Liver Disease Is Associated with an Increased Incidence of Atrial Fibrillation in Patients with Type 2 Diabetes. Clin. Sci. 2013, 125, 301–309.
- Hung, C.S.; Tseng, P.H.; Tu, C.H.; Chen, C.C.; Liao, W.C.; Lee, Y.C.; Chiu, H.M.; Lin, H.J.; Ho, Y.L.; Yang, W.S.; et al. Nonalcoholic Fatty Liver Disease Is Associated with QT Prolongation in the General Population. J. Am. Heart Assoc. 2015, 4, e001820.
- Mantovani, A.; Rigamonti, A.; Bonapace, S.; Bolzan, B.; Pernigo, M.; Morani, F.; Giovanni, L.; Bergamini, C.; Bertolini, L.; Valbusa, F.; et al. Nonalcoholic Fatty Liver Disease Is Associated with Ventricular Arrhythmias in Patients with Type 2 Diabetes Referred for Clinically Indicated 24-h Holter Monitoring. Diabetes Care 2016, 39, 1416–1423.
- Mantovani, A.; Rigolon, R.; Bonapace, S.; Morani, G.; Zoppini, G.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease Is Associated with an Increased Risk of Heart Block in Hospitalized Patients with Type 2 Diabetes Mellitus. PLoS ONE 2017, 12, e0185459.
- Tsang, T.S.; Barnes, M.E.; Miyasaka, Y.; Cha, S.S.; Bailey, K.R.; Verzosa, G.C.; Seward, J.B.; Gersh, B.J. Obesity as a Risk Factor for the Progression of Paroxysmal to Permanent Atrial Fibrillation: A Longitudinal Cohort Study of 21 Years. Eur. Heart J. 2008, 29, 2227–2233.
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Diabetologia 2016, 59, 1141–1144.
- Graner, M.; Nyman, K.; Siren, R.; Pentikainen, M.O.; Lundbom, J.; Hakkarainen, A.; Lauerma, K.; Lundbom, N.; Nieminen, M.S.; Taskinen, M.R. Ectopic Fat Depots and Left Ventricular Function in Nondiabetic Men with Nonalcoholic Fatty Liver Disease. Circ. Cardiovasc. Imaging 2014, 8, e001979.
- Käräjämäki, A.J.; Hukkanen, J.; Ukkola, O. The Association of Non-Alcoholic Fatty Liver Disease and Atrial Fibrillation: A Review. Ann. Med. 2018, 50, 371–380.
- Liu, Y.C.; Hung, C.S.; Wu, Y.W.; Lee, Y.C.; Lin, Y.H.; Lin, C.; Lo, M.T.; Chan, C.C.; Ma, H.P.; Ho, Y.L.; et al. Influence of Non-Alcoholic Fatty Liver Disease on Autonomic Changes Evaluated by the Time Domain, Frequency Domain, and Symbolic Dynamics of Heart Rate Variability. PLoS ONE 2013, 8, e61803.
- Park, H.W.; Shen, M.J.; Lin, S.F.; Fishbein, M.C.; Chen, L.S.; Chen, P.S. Neural Mechanisms of Atrial Fibrillation. Curr. Opin. Cardiol. 2012, 27, 24–28.
- Guo, Y.; Lip, G.Y.; Apostolakis, S. Inflammation in Atrial Fibrillation. J. Am. Coll. Cardiol. 2012, 60, 2263–2270.
- Ding, Y.H.; Ma, Y.; Qian, L.Y.; Xu, Q.; Wang, L.H.; Huang, D.S.; Zou, H. Linking Atrial Fibrillation with Non-Alcoholic Fatty Liver Disease: Potential Common Therapeutic Targets. Oncotarget 2017, 8, 60673–60683.
- Lin, Y.K.; Chen, Y.C.; Chen, J.H.; Chen, S.A.; Chen, Y.J. Adipocytes Modulate the Electrophysiology of Atrial Myocytes: Implications in Obesity-Induced Atrial Fibrillation. Basic Res. Cardiol. 2012, 107, 293.
- Chung, M.K.; Martin, D.O.; Sprecher, D.; Wazni, O.; Kanderian, A.; Carnes, C.A.; Bauer, J.A.; Tchou, P.J.; Niebauer, M.J.; Natale, A.; et al. C-Reactive Protein Elevation in Patients with Atrial Arrhythmias: Inflammatory Mechanisms and Persistence of Atrial Fibrillation. Circulation 2001, 104, 2886–2891.
- Mantovani, A.; Pernigo, M.; Bergamini, C.; Bonapace, S.; Lipari, P.; Valbusa, F.; Bertolini, L.; Zenari, L.; Pichiri, I.; Dauriz, M.; et al. Heart Valve Calcification in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Metabolism 2015, 64, 879–887.
- Mantovani, A. Nonalcoholic Fatty Liver Disease (NAFLD) and Risk of Cardiac Arrhythmias: A New Aspect of the Liver-Heart Axis. J. Clin. Transl. Hepatol. 2017, 5, 134–141.
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of Cardiovascular, Cardiac and Arrhythmic Complications in Patients with Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 1724–1745.
- Perseghin, G.; Lattuada, G.; De Cobelli, F.; Esposito, A.; Belloni, E.; Ntali, G.; Ragogna, F.; Canu, T.; Scifo, P.; Del Maschio, A.; et al. Increased Mediastinal Fat and Impaired Left Ventricular Energy Metabolism in Young Men with Newly Found Fatty Liver. Hepatology 2008, 47, 51–58.
- Camici, P.; Ferrannini, E.; Opie, L.H. Myocardial Metabolism in Ischemic Heart Disease: Basic Principles and Application to Imaging by Positron Emission Tomography. Prog. Cardiovasc. Dis. 1989, 32, 217–238.
- Taegtmeyer, H.; McNulty, P.; Young, M.E. Adaptation and Maladaptation of the Heart in Diabetes: Part I: General Concepts. Circulation 2002, 105, 1727–1733.
- Alp, H.; Karaarslan, S.; Eklioğlu, B.S.; Atabek, M.E.; Altın, H.; Baysal, T. Association between Nonalcoholic Fatty Liver Disease and Cardiovascular Risk in Obese Children and Adolescents. Can. J. Cardiol. 2013, 29, 1118–1125.
- De Simone, G.; Richard, B.D.; Marcello, C.; Mary, J.R.; Elisa, T.L.; Helaine, E.R.; Barbara, V.H. Metabolic Syndrome and Left Ventricular Hypertrophy in the Prediction of Cardiovascular Events-The Strong Heart Study. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 98–104.
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.B.; Marsala, J.; Meyer, T.; et al. Effect of Obesity and Insulin Resistance on Myocardial Substrate Metabolism and Efficiency in Young Women. Circulation 2004, 109, 2191–2196.
- Borges-Canha, M.; Neves, J.S.; Libâni, D.; Von-Hafe, M.; Vale, C.; Araújo-Martins, M.; Leite, A.R.; Pimentel-Nunes, P.; Davide, C.; Adelino, L.M. Association between Nonalcoholic Fatty Liver Disease and Cardiac Function and Structure—A Meta-Analysis. Endocrine 2019, 66, 467–476.
- Kocabay, G.; Karabay, C.Y.; Colak, Y.; Oduncu, V.; Kalayci, A.; Akgun, T.; Guler, A.; Kirma, C. Left Atrial Deformation Parameters in Patients with Non-Alcoholic Fatty Liver Disease: A 2D Speckle Tracking Imaging Study. Clin. Sci. 2014, 126, 297–304.
- Mondillo, S.; Cameli, M.; Caputo, M.L.; Lisi, M.; Palmerini, E.; Padeletti, M.; Ballo, P. Early Detection of Left Atrial Strain Abnormalities by Speckle-Tracking in Hypertensive and Diabetic Patients with Normal Left Atrial Size. J. Am. Soc. Echocardiogr. 2011, 24, 898–908.
- Bekler, A.; Gazi, E.; Erbag, G.; Binnetoglu, E.; Barutcu, A.; Sen, H.; Temiz, A.; Altun, B. Right Ventricular Function and Its Relationship with Grade of Hepatosteatosis in Non-Alcoholic Fatty Liver Disease. Cardiovasc. J. Afr. 2015, 26, 109–113.
- Anstee, Q.M.; Mantovani, A.; Tilg, H.; Targher, G. Risk of Cardiomyopathy and Cardiac Arrhythmias in Patients with Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 425–439.
- Volzke, H.; Haring, R.; Lorbeer, R.; Wallaschofski, H.; Reffelmann, T.; Empen, K.; Rettig, R.; John, U.; Felix, S.B.; Dorr, M. Heart Valve Sclerosis Predicts All-Cause and Cardiovascular Mortality. Atherosclerosis 2010, 209, 606–610.
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350.
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the Early Lesion of ‘Degenerative’ Valvular Aortic Stenosis. Histological and Immunohistochemical Studies. Circulation 1994, 90, 844–853.
- Goto, T.; Onuma, T.; Takebe, K.; Kral, J.G. The Influence of Fatty Liver on Insulin Clearance and Insulin Resistance in Non-Diabetic Japanese Subjects. Int. J. Obes. Relat. Metab. Disord. 1995, 19, 841–845.
- Paulista, M.; Marcello, R.; Baumeister, S.E.; Dörr, J.M.; Wallaschofski, H.; Völzke, H.; Lieb, W. Hepatic Steatosis Is Associated With Aortic Valve Sclerosis in the General Population The Study of Health in Pomerania (SHIP). Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1690–1695.
- Paulus, W.J.; Tschope, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271.
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin Signaling Coordinately Regulates Cardiac Size, Metabolism, and Contractile Protein Isoform Expression. J. Clin. Investig. 2002, 109, 629–639.
- Dhingra, R.; Gona, P.; Wang, T.J.; Fox, C.S.; D’Agostino, R.B.; Vasan, R.S. Serum Gamma-Glutamyl Transferase and Risk of Heart Failure in the Community. Arter. Thromb. Vasc. Biol. 2010, 30, 1855–1860.
- Wannamethee, S.G.; Whincup, P.H.; Shaper, A.G.; Lennon, L.; Sattar, N. Γ-Glutamyltransferase, Hepatic Enzymes, and Risk of Incident Heart Failure in Older Men. Arter. Thromb. Vasc. Biol. 2012, 32, 830–835.
- Emdin, M.; Pompella, A.; Paolicchi, A. γ-Glutamyltransferase, Atherosclerosis, and Cardiovascular Disease: Triggering Oxidative Stress within the Plaque. Circulation 2005, 112, 2078–2208.
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nagata, C.; Takeda, J.; Sarui, H.; Kawahito, Y.; Yoshida, N.; Suetsugu, A.; Kato, T.; et al. Nonalcoholic Fatty Liver Disease Is a Novel Predictor of Cardiovascular Disease. World J. Gastroenterol. 2007, 13, 1579–1584.
- Abdeldyem, S.M.; Goda, T.; Khodeir, S.A.; Abou, S.S.; Abd-Elsalam, S. Nonalcoholic Fatty Liver Disease in Patients with Acute Ischemic Stroke Is Associated with More Severe Stroke and Worse Outcome. J. Clin. Lipidol. 2017, 11, 915–919.
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Cardio-Metabolic Disorders in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 2215.
- Bots, M.L.; Salonen, J.; Elwood, P.; Nikitin, Y.; Freire, D.; Inzitari, D.; Sivenius, J.; Trichopoulou, A.; Tuomilehto, J.; Koudstaal, P.; et al. Gamma-Glutamyltransferase and Risk of Stroke: The EUROSTROKE Project. J. Epidemiol. Community Health 2002, 56, i25–i29.
- Paolicchi, A.; Emdin, M.; Ghliozeni, E.; Ciancia, E.; Passino, C.; Popoff, G.; Pompella, A. Images in Cardiovascular Medicine, Human Atherosclerotic Plaques Contain Gamma-Glutamyl Transpeptidase Enzyme Activity. Circulation 2004, 109, 1440.
- Emdin, M.; Passino, C.; Franzini, M.; Paolicchi, A.; Pompella, A. γ-Glutamyltransferase and Pathogenesis of Cardiovascular Diseases. Future Cardiol. 2007, 263–270.
- Khalaf, M.R.; Hayhoe, F.G. Cytochemistry of Gamma-Glutamyltransferase in Haemic Cells and Malignancies. Histochem. J. 1987, 19, 385–395.
- Stark, A.A.; Zeiger, E.; Pagano, D.A. Glutathione Metabolism by Gamma-Glutamyltranspeptidase Leads to Lipid Peroxidation: Characterization of the System and Relevance to Hepatocarcinogenesis. Carcinogenesis 1993, 14, 183–189.
- Stocker, R.; Keaney, J.; John, F. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478.
- Ciavarella, A.; Gnocchi, D.; Custodero, C.; Lenato, G.M.; Fiore, G.; Sabbà, C.; Mazzocca, A. Translational Insight into Prothrombotic State and Hypercoagulation in Nonalcoholic Fatty Liver Disease. Thromb. Res. 2021, 198, 139–150.
- Di Minno, M.N.D.; Tufano, A.; Rusolillo, A.; Di Minno, G.; Tarantino, G. High Prevalence of Nonalcoholic Fatty Liver in Patients with Idiopathic Venous Thromboembolism. World J. Gastroenterol. 2010, 28, 6119–6122.
- Stine, J.; Jonathan, G.; Shah, N.L.; Argo, C.K.; Pelletier, S.J.; Caldwell, S.H.; Northup, P.G. Northup Increased Risk of Portal Vein Thrombosis in Patients With Cirrhosis Due to Nonalcoholic Steatohepatitis. Liver Transpl. 2015, 21, 1016–1021.
- Nieuwdorp, M.; Stroes, E.S.; Meijers, J.C.; Buller, H. Hypercoagulability in the Metabolic Syndrome. Curr. Opin. Pharmacol. 2005, 5, 155–159.
- Kim, J.A.; Kim, J.E.; Song, S.H.; Kim, H.K. Influence of Blood Lipids on Global Coagulation Test Results. Ann. Lab. Med. 2015, 35, 15–21.
- Yun, J.W.; Cho, Y.K.; Park, J.H.; Kim, H.J.; Park, D.I.; Sohn, C.I.; Jeon, W.K.; Kim, B.I. Abnormal Glucose Tolerance in Young Male Patients with Nonalcoholic Fatty Liver Disease. Liver Int. 2009, 29, 525–529.
- Undas, A.; Wiek, I.; Stepien, E.; Zmudka, K.; Tracz, W. Hyperglycemia Is Associated with Enhanced Thrombin Formation, Platelet Activation, and Fibrin Clot Resistance to Lysis in Patients with Acute Coronary Syndrome. Diabetes Care 2008, 31, 1590–1595.
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet Dysfunction in Type 2 Diabetes. Diabetes Care 2001, 24, 1476–1485.
- Ceriello, A. Coagulation Activation in Diabetes Mellitus: The Role of Hyperglycaemia and Therapeutic Prospects. Diabetologia 1993, 36, 1119–1125.
- Mitropoulos, K.A.; Miller, G.J.; Watts, G.F.; Durrington, P.N. Lipolysis of Triglyceride-Rich Lipoproteins Activates Coagulant Factor XII: A Study in Familial Lipoprotein-Lipase Deficiency. Atherosclerosis 1992, 95, 119–125.
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem. J. 2001, 357, 593–615.
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic Profiling Reveals a Contribution of Gut Microbiota to Fatty Liver Phenotype in Insulin-Resistant Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516.
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of Gut-Flora-Dependent Metabolite Trimethylamine-Noxide, Betaine and Choline with Non-Alcoholic Fatty Liver Disease in Adults. Sci. Rep. 2016, 6, 19076.
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124.
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; Martino, J.S.; Pirola, C.J. Circulating Levels and Hepatic Expression of Molecular Mediators of Atherosclerosis in Nonalcoholic Fatty Liver Disease. Atherosclerosis 2010, 209, 585–591.
- Esmon, C.T. The Interactions between Inflammation and Coagulation. Br. J. Haematol. 2005, 131, 417–430.
- Dole, V.S.; Bergmeier, W.; Patten, I.S.; Hirahashi, J.; Mayadas, T.N.; Wagner, D.D. PSGL-1 Regulates Platelet P-Selectin-Mediated Endothelial Activation and Shedding of P-Selectin from Activated Platelets. Thromb. Haemost. 2007, 98, 806–812.


