- Subjects: Agriculture, Dairy & Animal Science
- |
- Contributors:
- Diamanto Skopelitou ,
- Beiping Miao ,
- Abhishek Kumar ,
- Dagmara Dymerska ,
- Jan Lubiński ,
- Kari Hemminki ,
- Asta Försti
- familial colorectal cancer
- whole exome sequencing
- germline variant
- HDAC5
- APCDD1
- 5'UTR
- promoter activity
This video is adapted from 10.3390/ijms23031295
1. Current status of genetic causes in familial colorectal cancer and aims of the presented work
Colorectal cancer (CRC) has one of the highest proportions of familial cases among hereditary malignancies, and heritable factors are estimated to account for about 35% of CRC risk according to twin studies[1]. Traditional cancer-predisposing germline mutations in APC, MUTYH and mismatch repair genes (MLH1, MSH2, MSH6, PMS2 and EPCAM) are already known to be associated with familial CRC and lead to phenotypes of well-defined Mendelian CRC syndromes, such as familial adenomatous polyposis (FAP); MUTYH-associated polyposis (MAP); and Lynch syndrome, a hereditary non-polyposis colon cancer (HNPCC) syndrome, respectively[2]. Recent sequencing studies have further proposed several novel CRC predisposition genes in recent years: germline variants in HNRNPA0 and WIF1 genes have been identified in a family with susceptibility to multiple early onset cancers including CRC[3], while a germline mutation in NTHL1 gene has been found in three unrelated families with adenomatous polyposis and various cancer types including CRC[4][5]. Moreover, both germline and somatic mutations in the POLE and POLD1 genes have been associated with both sporadic and familial CRC, contributing to the genetic understanding of CRC inheritance[6][7][8]. Nevertheless, germline mutations in these or other established predisposition genes account for only 5 - 10% of all CRC cases[9], leaving the genetic background of most familial CRC cases as still not sufficiently explored. Thus, the application of next generation sequencing techniques on these patients in the context of pedigree-based studies bears great potential for exploring the remaining genetic burden of familial CRC.
2. Whole exome sequencing and variant evaluation
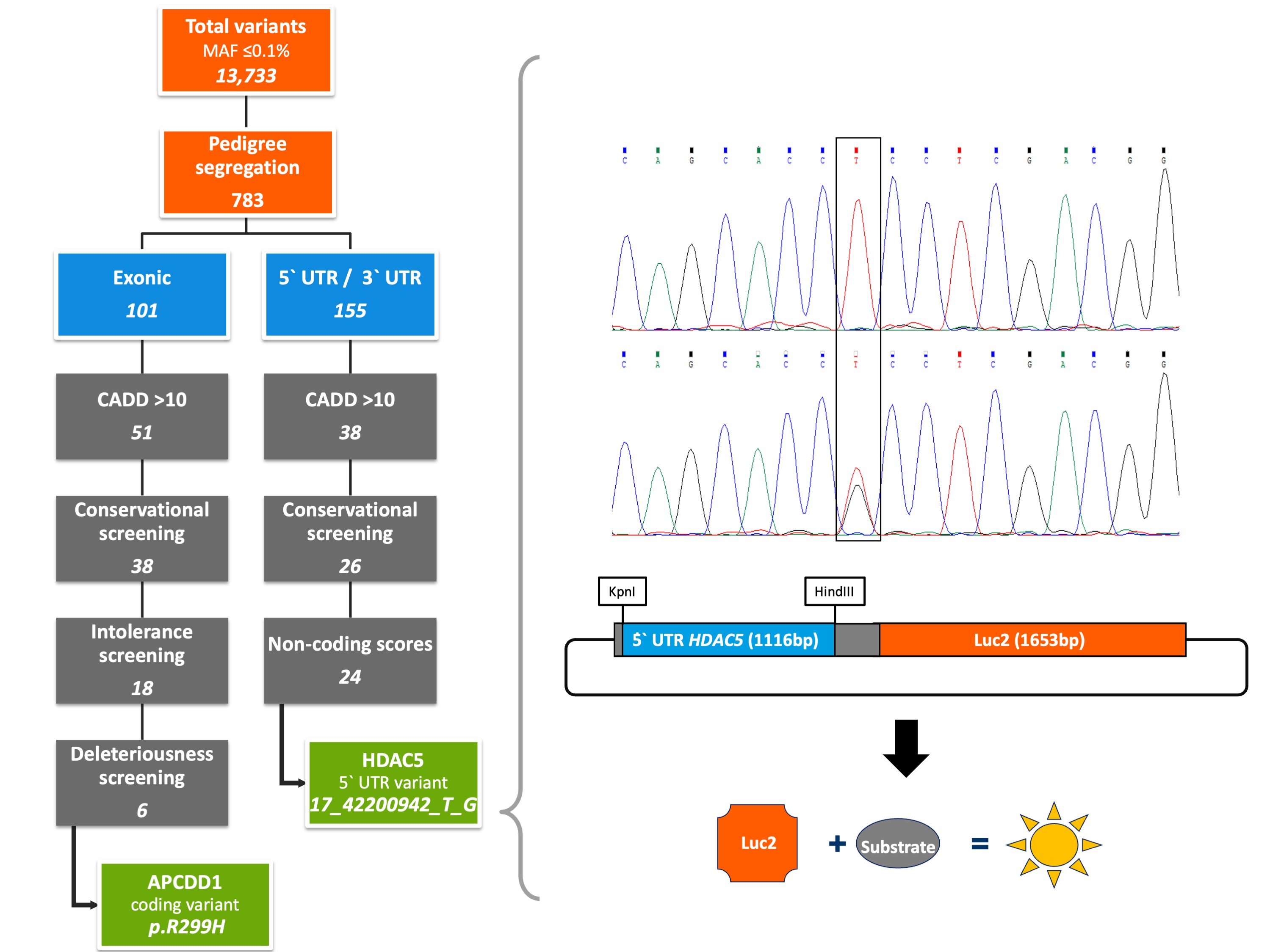
With the aim of identifying rare high- to moderate-penetrance germline variants underlying CRC susceptibility, we performed whole exome sequencing on three CRC cases and three unaffected members of a Polish family with a history of CRC over three generations. We identified a total of 13,733 variants with a minor allele frequency of ≤ 0.1% and passed the resulting data through our in-house developed Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2). Based on evolutionary conservation, intolerance to genetic variation, overall deleteriousness, and the presence of regulatory elements, we evaluated both coding and non-coding variants. Evolutionary conservation was assessed using Genomic Evolutionary Rate Profiling[10], PhastCons[11] and PhyloP scores[12], whereas the applied intolerance scores were based on allele frequency data from our in-house datasets, ESP[13] and Exome Aggregation Consortium (ExAC)[14]. Deleteriousness scoring tools were accessed from dbNSFP v3.0 (database for nonsynonymous SNVs' functional predictions)[15]. Variants potentially affecting regulatory elements, such as binding motifs in promoters, enhancers or super-enhancers, were identified using the Combined Annotation Dependent Depletion (CADD) tool version 1.4[16], SNPnexus[17] and the intersect function of Bedtools applied to datasets derived from the FANTOM5 consortium[18] and the Super-Enhancer Archive[19].
Our integrative analysis resulted in the prioritization of two novel heterozygous variants: a coding variant in exon 4 of the APC downregulated 1 gene (APCDD1) that induces the amino acid substitution R299H in the second of two functional APCDDC domains and a non-coding variant in the 5′ untranslated region (UTR) of the histone deacetylase 5 gene (HDAC5) located about 6 kb upstream of the transcription start site (17_42200942_T_G). Whereas the APCDD1 gene is known to be involved in the Wnt signaling pathway as a direct target of the beta-Catenin/TCF4 complex[20], the HDAC5 gene has been implicated in colorectal carcinogenesis by upregulating the Delta-like 4 ligand (DLL4), a vascular-specific Notch ligand essential for tumor angiogenesis[21][22]. Familial segregation of both identified variants with the disease was verified by the Integrative Genomics Viewer, a visualization tool for interactive exploration of large genomic datasets[23], and further confirmed by targeted Sanger sequencing.
3. Screening of large case and control cohorts and functional in vitro experiments
In order to assess the functional impact of the identified variants in APCDD1 and HDAC5 genes, we screened a large cohort of familial CRC patients using Taqman assays and performed further functional experiments in vitro:
Taqman assays for the R299H variant in the APCDD1 gene, revealed 8 additional variants in a cohort of 1,705 familial CRC patients and 2 findings in a cohort of 1,674 healthy elderly individuals from Poland (OR = 4.44, 95% CI = [0.96; 20.56], p = 0.06). On the other hand, cell proliferation assays using human embryonic kidney cells (HEK293T) and human colon cancer cells (HT-29) showed an insignificant proliferative impact for the variant, excluding the APCDD1 variant as the sole potential cancer-predisposing candidate in the studied family.
No further individual carrying the 5′UTR variant was found in either the screened cohort of 1,705 familial CRC patients or the control cohort of 1,674 healthy elderly individuals. Nevertheless, functional experiments showed a significant impact of the identified non-coding variant on HDAC5 expression. In order to investigate whether the identified variant may affect transcriptional regulation of HDAC5 gene expression, we cloned gene reporter constructs including the mutated or wild-type sequence of the 5’ UTR of HDAC5 gene and performed luciferase reporter assays using HEK293T cells. The results of our experiments showed significantly enhanced promoter activity of the HDAC5 gene carrying the non-coding variant compared to the wild-type sequence. Since in silico analyses using Jaspar 2020[24] predicted the disruption of multiple transcription factor binding sites (TFBS) by the identified 5’ UTR variant, we performed further luciferase reporter assays targeting such TFBS affected by the mutation. After co-expression of the transcription factor TCF4, involved in the Wnt signaling pathway and reported as a negative prognostic factor in CRC[25], a further upregulation of promoter activity was observed especially in cells carrying the mutation. Hence, the results of our experiments implicated TCF4 as a potential regulator of gene expression of mutated HDAC5, thus contributing to colorectal carcinogenesis in the studied family.
4. Conclusion and future directions
By identifying germline variants in APCDD1 and HDAC5 genes in a family with CRC history over three generations, we hope to not only facilitate the exploration of unrecognized genetic causes of familial CRC but also to underline the relevance of non-coding variants in the 5’ UTR that affect transcriptional regulation and play a role in the pathogenesis of complex disorders. In conclusion, our findings emphasize the importance of considering both coding and non-coding variants in the genetic research of familial cancers.
- Paul Lichtenstein; Niels Vilstrup Holm; Pia K. Verkasalo; Anastasia Iliadou; Jaakko Kaprio; Markku Koskenvuo; Eero Pukkala; Axel Skytthe; Kari Hemminki; Environmental and Heritable Factors in the Causation of Cancer — Analyses of Cohorts of Twins from Sweden, Denmark, and Finland. New England Journal of Medicine 2000, 343, 78-85, 10.1056/nejm200007133430201.
- Kory W. Jasperson; Thérèse M. Tuohy; Deborah W. Neklason; Randall W. Burt; Hereditary and Familial Colon Cancer. Gastroenterology 2010, 138, 2044-2058, 10.1053/j.gastro.2010.01.054.
- Chongjuan Wei; Bo Peng; Younghun Han; Wei V. Chen; Joshua Rother; Gail E. Tomlinson; C. Richard Boland; Marc Chaussabel; Marsha L. Frazier; Christopher I. Amos; et al. Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Familial Cancer 2015, 14, 297-306, 10.1007/s10689-014-9758-8.
- Roland P. Kuiper; Nicoline Hoogerbrugge; NTHL1 defines novel cancer syndrome. Oncotarget 2015, 6, 34069-34070, 10.18632/oncotarget.5864.
- Robbert D A Weren; Marjolijn J L Ligtenberg; C Marleen Kets; Richarda M De Voer; Eugène T P Verwiel; Liesbeth Spruijt; Wendy A G Van Zelst-Stams; Marjolijn C Jongmans; Christian Gilissen; Jayne Y Hehir-Kwa; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nature Genetics 2015, 47, 668-671, 10.1038/ng.3287.
- Sarah Briggs; Ian Tomlinson; Germline and somatic polymerase ϵ and δ mutations define a new class of hypermutated colorectal and endometrial cancers. The Journal of Pathology 2013, 230, 148-153, 10.1002/path.4185.
- Claire Palles; Jean-Baptiste Cazier; Kimberley M. Howarth; Enric Domingo; Angela M. Jones; Peter Broderick; Zoe Kemp; Sarah L. Spain; Estrella Guarino; Israel Salguero; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genetics 2012, 45, 136-144, 10.1038/ng.2503.
- Laura Valle; Richarda M. de Voer; Yael Goldberg; Wenche Sjursen; Asta Försti; Clara Ruiz-Ponte; Trinidad Caldés; Pilar Garré; Maren F. Olsen; Margareta Nordling; et al. Update on genetic predisposition to colorectal cancer and polyposis. Molecular Aspects of Medicine 2019, 69, 10-26, 10.1016/j.mam.2019.03.001.
- Marie Lorans; Eryn Dow; Finlay A. Macrae; Ingrid M. Winship; Daniel D. Buchanan; Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clinical Colorectal Cancer 2018, 17, e293-e305, 10.1016/j.clcc.2018.01.001.
- Gregory M. Cooper; Eric A. Stone; George Asimenos; Eric D. Green; Serafim Batzoglou; Arend Sidow; Distribution and intensity of constraint in mammalian genomic sequence. Genome Research 2005, 15, 901-913, 10.1101/gr.3577405.
- Adam Siepel; Gill Bejerano; Jakob S. Pedersen; Angie S. Hinrichs; Minmei Hou; Kate Rosenbloom; Hiram Clawson; John Spieth; LaDeana W. Hillier; Stephen Richards; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Research 2005, 15, 1034-1050, 10.1101/gr.3715005.
- Katherine S. Pollard; Melissa J. Hubisz; Kate R. Rosenbloom; Adam Siepel; Detection of nonneutral substitution rates on mammalian phylogenies. Genome Research 2009, 20, 110-121, 10.1101/gr.097857.109.
- Slavé Petrovski; Quanli Wang; Erin L. Heinzen; Andrew S. Allen; David B. Goldstein; Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLOS Genetics 2013, 9, e1003709, 10.1371/journal.pgen.1003709.
- Monkol Lek; Exome Aggregation Consortium; Konrad J. Karczewski; Eric V. Minikel; Kaitlin E. Samocha; Eric Banks; Timothy Fennell; Anne H. O’Donnell-Luria; James S. Ware; Andrew J. Hill; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285-291, 10.1038/nature19057.
- XiaoMing Liu; Chunlei Wu; Chang Li; Eric Boerwinkle; dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Human Mutation 2015, 37, 235-241, 10.1002/humu.22932.
- Philipp Rentzsch; Daniela Witten; Gregory M Cooper; Jay Shendure; Martin Kircher; CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research 2018, 47, D886-D894, 10.1093/nar/gky1016.
- Abu Z Dayem Ullah; Jorge Oscanoa; Jun Wang; Ai Nagano; Nicholas R Lemoine; Claude Chelala; SNPnexus: assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Research 2018, 46, W109-W113, 10.1093/nar/gky399.
- Marina Lizio; The FANTOM consortium; Jayson Harshbarger; Hisashi Shimoji; Jessica Severin; Takeya Kasukawa; Serkan Sahin; Imad Abugessaisa; Shiro Fukuda; Fumi Hori; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biology 2015, 16, 1-14, 10.1186/s13059-014-0560-6.
- Yanjun Wei; Shumei Zhang; Shipeng Shang; Bin Zhang; Song Li; Xinyu Wang; Fang Wang; Jianzhong Su; Qiong Wu; Hongbo Liu; et al. SEA: a super-enhancer archive. Nucleic Acids Research 2015, 44, D172-D179, 10.1093/nar/gkv1243.
- Meiko Takahashi; Manabu Fujita; Yoichi Furukawa; Ryuji Hamamoto; Takashi Shimokawa; Nobutomo Miwa; Michio Ogawa; Yusuke Nakamura; Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis.. Cancer Research 2002, 62, 5651–5656, .
- Marina Badenes; Alexandre Trindade; Hugo Pissarra; Luís Lopes-Da-Costa; António Duarte; Delta-like 4/Notch signaling promotes Apc Min/+ tumor initiation through angiogenic and non-angiogenic related mechanisms.. BMC Cancer 2017, 17, 50, 10.1186/s12885-016-3036-0.
- Ping He; Jiexiong Liang; Tiansong Shao; Yang Guo; Yingchen Hou; Yang Li; HDAC5 promotes colorectal cancer cell proliferation by up-regulating DLL4 expression. International journal of clinical and experimental medicine 2015, 8, 6510-6516, .
- James T. Robinson; Helga Thorvaldsdóttir; Aaron M. Wenger; Ahmet Zehir; Jill P. Mesirov; Variant Review with the Integrative Genomics Viewer. Cancer Research 2017, 77, e31-e34, 10.1158/0008-5472.can-17-0337.
- Oriol Fornes; Jaime A Castro-Mondragon; Aziz Khan; Robin Van Der Lee; Xi Zhang; Phillip A Richmond; Bhavi P Modi; Solenne Correard; Marius Gheorghe; Damir Baranašić; et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Research 2019, 48, D87-D92, 10.1093/nar/gkz1001.
- Lydia Kriegl; David Horst; Jana A Reiche; Jutta Engel; Thomas Kirchner; Andreas Jung; LEF-1 and TCF4 expression correlate inversely with survival in colorectal cancer. Journal of Translational Medicine 2010, 8, 123-123, 10.1186/1479-5876-8-123.