Fibrinolysis is a complex enzymatic process aimed at dissolving blood clots to prevent vascular occlusions. The fibrinolytic system is composed of a number of cofactors that, by regulating fibrin degradation, maintain the hemostatic balance. A dysregulation of fibrinolysis is associated with various pathological processes that result, depending on the type of abnormality, in prothrombotic or hemorrhagic states.
1. Introduction
Fibrinolysis is a delicate and complex enzymatic process aimed at dissolving blood clots, thereby localizing and limiting clot formation
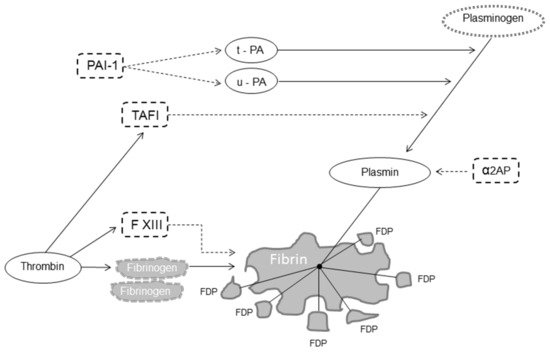
[1][2][3]. Fibrinolysis is responsible for fibrin degradation and is modulated by proteases and protease inhibitors that, having the opposite effect, regulate the conversion of plasminogen to plasmin, the active enzyme that dissolves the fibrin clot into soluble fibrin degradation products (FDP)
[4]. The fibrinolytic system thus includes pro-fibrinolytic (i.e., tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA)) and anti-fibrinolytic components (i.e., α2-antiplasmin, plasminogen activator inhibitor 1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI)) (
Figure 1)
[5].
Figure 1. Fibrinolysis. Legend: dot arrow inhibits fibrinolysis; solid line favors fibrinolysis. Abbreviations: FXIII, factor XIII, t-PA, tissue plasminogen activator; u-PA, urokinase plasminogen activator; α2AP, α2-antiplasmin; PAI-1, plasminogen activator inhibitor 1; TAFI, thrombin-activatable fibrinolysis inhibitor; FDP, fibrin degradation products.
While under physiological conditions, fibrinolysis (also called primary fibrinolysis) is a state of balance between these two opposite modulators; an abnormally reduced or excessive fibrinolysis disrupts the hemostatic balance and can lead to thrombosis or clinical bleeding, respectively
[6]. Conditions causing a hypo- or hyper-fibrinolytic state are inherited, typically caused by a single molecular defect or more frequently acquired
[6][7].
2. Inherited Bleeding Disorders of Primary Fibrinolysis
The inherited bleeding disorders of primary fibrinolysis include α2-antiplasmin deficiency, plasminogen activator inhibitor-1 deficiency and the Quebec platelet disorder.
2.1. α2-Antiplasmin Deficiency
The α
2-plasmin inhibitor, a single-chain glycoprotein of 70 kDa synthesized by the liver, is the primary physiological inhibitor of plasmin
[8]. Congenital α
2-antiplasmin deficiency is a rare autosomal recessive condition (the gene is mapped to chromosome 17) characterized by clinical bleeding due to premature dissolution of hemostatic plugs, typically presenting as re-bleeding following trauma or invasive/surgical procedures
[9][10]. α
2-plasmin inhibits fibrinolysis by forming a stable inactive complex with plasmin by binding to the lysine-binding site of plasminogen and thus completely inhibiting plasminogen binding to fibrin, and by covalent binding to fibrin via activated factor XIII (FXIIIa) to prevent fibrinolysis by plasmin
[1][11].
The diagnosis of α
2-antiplasmin deficiency is difficult because coagulation screening tests are normal, but the euglobulin clot lysis time (ECLT) is short due to uninhibited plasmin activity. The functional and immunologic plasma assays of the inhibitor confirm the diagnosis
[10]. Several mutations have been identified in the gene encoding α
2-antiplasmin that cause two different types of α
2-antiplasmin deficiency: a quantitative defect (type I), characterized by a similar decrease in both antigen and activity plasma levels, and a qualitative defect (type II), characterized by lower functional activity contrasting with normal antigen levels
[10].
Case reports of both children and adults with congenital deficiency of α
2-plasmin inhibitor
[12][13][14][15][16] include a wide array of bleeding manifestations, such as umbilical cord bleeding, prolonged bleeding from wounds, epistaxis, gingival bleeding, subcutaneous and intramuscular hematomas, hematuria, hemarthroses and central nervous system bleeding. However, only homozygotes (presenting with α
2-plasmin inhibitor levels < 10%) experience a significant bleeding tendency, usually severe and starting from childhood, while in heterozygotes (with levels between 20% and 50%), hemorrhagic events are reported to occur only after trauma, dental extractions or surgery
[10]. The use of the antifibrinolytic agent tranexamic acid (TXA) is effective for preventing bleeding during or after invasive/surgical procedures and treating acute bleeding episodes. Fresh-frozen plasma (FFP) may be used as an alternative or an adjunct to antifibrinolytic therapy when an immediate increase of this fibrinolysis inhibitor is required
[17]. Solvent–detergent treated plasma has lower α2-antiplasmin levels and thus is not recommended in the treatment of α2-antiplasmin deficiency
[7].
2.2. Plasminogen Activator Inhibitor-1 Deficiency
PAI-1 is a single-chain glycoprotein of 52 kDa synthesized in the liver (the
PAI-1 gene is located at chromosome 7), which controls the proteolytic action of plasmin through inhibition of the plasminogen activators t-PA and u-PA
[18][19][20]. Congenital PAI-1 deficiency, transmitted as an autosomal recessive trait, may be quantitative (decreased or absent protein production) or qualitative (dysfunctional PAI-1 synthesis with detectable protein but reduced or absent functional activity)
[19]. As for α
2-antiplasmin deficiency, the diagnosis of congenital PAI-1 deficiency is challenging because screening coagulation tests are normal. Although the ECLT is short and whole blood clotting assays such as thromboelastogram are abnormal, diagnosis is based on the results of antigenic (enzyme-linked immunosorbent assay, ELISA) and functional (chromogenic test) PAI-1 assays
[19]. Congenital PAI-1 deficiency is an extremely rare bleeding disorder. While heterozygotes usually have no bleeding manifestations, PAI-1 deficiency in homozygotes appears to be a mild to moderate bleeding disorder
[21][22][23][24][25]. These patients rarely have spontaneous bleeding but experience abnormal bleeding only following trauma or surgery. The bleeding tendency from congenital PAI-1 deficiency was also displayed in infancy
[19] and children
[24]. Menorrhagia and obstetric complications (miscarriages and preterm births) have been reported in PAI-1 deficient females
[26]. Antifibrinolytic agents such as TXA are the mainstay of treatment of acute bleeding. The prophylactic administration of this drug is also very effective in preventing bleeding in patients undergoing surgery, especially oral and urogenital procedures. Persistent menorrhagia may be effectively managed with hormonal therapy and long-term prophylactic antifibrinolytic therapy if necessary. TXA is used at typical doses of 25 mg/kg body weight every 8 h. The duration of treatment for prevention and treating bleeds is usually 3–4 days for minor bleeds/surgeries and 5–7 days for major bleeds/surgeries. Desmopressin acetate should be avoided as it may induce endothelial secretion of plasminogen activator
[7].
2.3. Quebec Platelet Disorder
The Quebec platelet syndrome is a rare autosomal dominant bleeding disorder associated with a mildly reduced platelet count and caused by overexpression of u-PA and its increased storage in platelet α-granules, leading to plasmin-mediated α-granule protein degradation, increased release of u-PA and hyperfibrinolysis
[27][28][29]. The candidate gene
PLAU (urokinase plasminogen activator) is mapped on chromosome 10
[30]. The laboratory characteristic is a marked increase of platelet content of the u-PA. From a clinical point of view, patients with the Quebec platelet syndrome typically present with delayed bleeding following trauma or surgical and dental procedures, but can also experience easy bruising, epistaxis, hematuria, menorrhagia and joint bleeds
[31]. Sometimes bleeding may be severe and uncontrolled despite therapy. The mainstay of prevention and treatment of bleeding is based on antifibrinolytic agents, which should be avoided in patients with hematuria due to the risk of urinary tract clot formation leading to renal obstruction
[28]. Prophylaxis with fibrinolytic inhibitors has also been successfully used during childbirth
[31]. Platelet transfusions have been used but may not be beneficial in the case of severe bleeding
[29].
3. Acquired Bleeding Disorders of Primary Fibrinolysis
The acquired bleeding disorders of primary fibrinolysis include liver cirrhosis, acute promyelocytic leukemia, severe trauma and post-partum hemorrhage.
3.1. Liver Cirrhosis
Severe liver disease is frequently complicated by clinically significant bleeding due to multiple causes, including endothelial dysfunction and thrombocytopenia caused by splenic sequestration, portal hypertension with the development of varices and a de-creased synthesis of coagulation factors and their inhibitors associated with impaired hepatic function
[32][33][34].
A hyperfibrinolytic state is a common finding in liver cirrhosis and may
[35][36][37] occur in up to 50% of patients with end-stage liver disease and is associated with mucocutaneous and gastrointestinal bleeding
[38]. In this clinical setting, the fibrinolytic system is strongly activated by an increased endothelial release and decreased hepatic clearance of t-PA, decreased synthesis of TAFI, α
2-plasmin inhibitor and PAI-1
[39][40][41][42]. In a study of 112 patients with liver cirrhosis with esophageal varices but without upper gastrointestinal bleeding followed for 3 years, elevated t-PA and D-dimer levels were the only markers predictive of bleeding
[43]. The successful use of antifibrinolytic agents in patients with cirrhosis, particularly those with mucosal or gastrointestinal bleeding, further supports the mechanistic role of primary hyperfibrinolysis in this clinical setting
[32].
3.2. Acute Promyelocytic Leukemia
Acute promyelocytic leukemia (APL) has peculiar molecular, morphological and clinical characteristics that render this type of acute myeloid leukemia unique
[44]. A single genetic defect has been identified in APL, a translocation between chromosomes 15 and 17 resulting in the fusion of the promyelocytic leukemia protein gene (
PML) and the retinoic acid receptor-α gene (
RARA). The expressed PML-RAR-α fusion protein stops the differentiation of myeloid precursor cells and prolongs their survival, resulting in APL
[45]. APL is characterized by an increased incidence of severe bleeding complications
[44]. The hemostatic abnormalities in APL have the laboratory features of primary hyperfibrinolysis (i.e., elevated circulating levels of u-PA and t-PA and reduced levels of PAI-1, α2-antiplasmin and TAFI), and the responsible molecular mechanism has been elucidated in the last decade
[46][47][48][49][50][51][52]. The fusion protein PML-RAR-enhances the expression of the S100 protein (a member of the S100 family of calcium-binding proteins), which in turn forms a heterotetrameric complex with annexin A2, a protein receptor with a strong affinity for plasminogen and t-PA as well as a potent cofactor for the conversion of plasminogen to plasmin. Thus, the surface-bound S100-annexin A2 complex promotes plasminogen activation and, at the same time, protects plasmin from inhibition
[53]. Notably, the expression of annexin A2 can also be directly enhanced by PML-RAR-α
[54].
3.3. Trauma
Trauma is a leading global health burden, being responsible for over 6 million deaths worldwide each year
[55]. Notably, nearly 50% of trauma deaths result from bleeding complications and occur in the first few hours after trauma
[55].
The precise mechanism underlying fibrinolysis activation in severe trauma is not completely elucidated, although the disruption of fibrinogen metabolism induced by hypothermia and metabolic acidosis and the resulting direct fibrinogenolysis have been indicated as potential mechanisms
[56]. In addition, the activation of the protein C pathway seems to play a central role in enhancing systemic fibrinolysis and inducing fibrinogen depletion
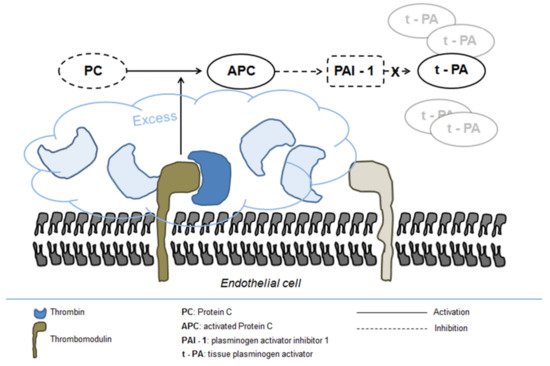
[55]. Protein C activation is markedly enhanced following trauma owing to the massive generation of thrombin and its complex formation with thrombomodulin on the endothelial surface
[56]. Excess activated protein C in turn inhibits PAI-1 release leading to increased concentration of tPA (
Figure 2)
[57].
Figure 2. Fibrinolysis activation in severe trauma.
3.4. Post-Partum Hemorrhage
Post-partum hemorrhage, a major cause of maternal death worldwide, is commonly defined as blood loss of 500 mL or more within 24 h and is characterized by the early onset of hyperfibrinolysis
[58][59]. The pathogenesis of increased fibrinolytic activity is poorly understood, but it seems to be similar (i.e., increased endothelial t-PA production and inhibition of PAI-1 via protein C activation) to that described in severe trauma
[60]. In physiological pregnancies, FPD levels raise in the hours following delivery, but this increase is many times higher in women with post-partum hemorrhage
[59]. The significant role played by fibrinolysis in post-partum hemorrhage is further supported by the recent finding of increased D-dimer levels and plasmin-antiplasmin complexes in the immediate post-partum period
[61].
 +1 point
+1 point