Early diagnosis of AKI in the setting of sepsis is important in order to provide optimal treatment and avoid further kidney injury
[48]. Therefore, the use of injury or stress biomarkers, in addition to the clinical assessment of renal function, is required
[49][50]. Inflammation appears to play an important role in sepsis-related kidney injury. Interleukin-6 (IL-6) has been described as predictive of AKI, independently of hypotension (e.g., mean arterial pressure, dosage of vasopressors)
[51].
Since HDL levels significantly declined in severe sepsis and septic shock, several studies evaluated HDL impact on renal damage as a possible predictive biomarker and therapeutic target
[52]. In a blinded, observational cohort study, the authors analyzed HDL amount in 200 adult patients with suspected sepsis at the time of admission
[12]. They observed that HDL concentration is a prognostic factor to establish an early efficient therapy to avoid disease progression, multi-organ dysfunction, and renal damage
[12]. In another observational small study of 180 patients with clear signs of infections, the authors demonstrated that low HDL levels are associated with increased risk of AKI onset and decreased glomerular filtration rate (GFR)
[16]. These results suggest that HDL levels may accurately predict the development and the stages of AKI like serum creatinine levels for the Kidney Disease Improving Global Outcomes (KDIGO) index
[16]. Interestingly, low levels of HDL in the early phase of sepsis could represent predictors of worsening renal function at three to twenty-four months after hospital discharge of septic patients with a dyslipidemia profile
[16]. These results underline the key role of HDL in affecting renal function and disease progression.
Most of the current understanding of HDL involvement in sepsis pathogenesis and AKI have been revealed from animal models and in vitro studies. For example, Guo L. et al. demonstrated that higher expression of ApoA-I in transgenic mice increased survival and limited sepsis and renal damage compared to wild type mice
[8][9]. HDL might provide renal protection through several mechanisms including pathogens detoxification via SR-BI internalization. As discussed above, SR-BI is expressed by immune cells, endothelial cells, and parenchymal cells such as hepatocytes and renal tubular cells and mediates the cholesterol efflux to circulating HDL. In addition, pathogens molecules as LPS and LTA could bind HDL in the bloodstream and could be transferred to SR-BI for hepatic clearance. Decreased serum levels of LPS or LTA induced lower activation of TLR-4 and TLR-2 respectively, in renal parenchymal cells
[7][54][55][56]. Since both TLR-4 ad TLR-2 activation is associated with increased renal damage
[57][58], it is obviously that HDL levels ameliorate renal function, avoiding tubular damage, interstitial inflammation, and cytokines release.
Endothelial dysfunction is a hallmark of sepsis-associated AKI
[59]. In inflammatory conditions, endothelial cells increased inducible nitric oxide synthase activity (iNOS) and decreased eNOS activation
[60], leading to vascular impairment and renal parenchymal damage
[59]. Increased levels of HDL could activate the eNOS pathway, reducing adhesion molecules expression and leucocytes activation and neutrophil infiltration, assuring reduced renal damage
[61].
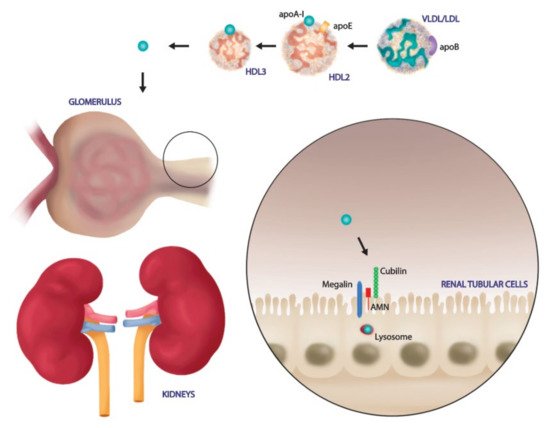
Renal tubular cells are not only the principal target of renal dysfunction, but they are directly involved in HDL modulation
[62]. In the physiological condition, the glomerular filtration barrier is intact and prevents passage of large molecules such as mature HDL. Only lipid poor ApoA-I, ApoA-I, ApoA-IV and enzymes such as LCAT cross the filtration barrier. Then, renal tubular cells express on their surface membrane specific receptors to bind and internalize these particles. In particular, pre-β HDL and lipid poor ApoA-I bind to the cubulin amnion less complex on the surface of renal proximal tubule cells and are endocytosed; this process is accelerated by the binding of another membrane protein, megalin, that is essential for renal proximal tubule reabsorption
[62]. After endocytosis, HDL could be degraded in tubular lysosome or transcytosed into intratubular lumen in order to be released into systemic circulation ().
Moreover, kidneys can also affect HDL metabolism, influencing extrarenal synthesis and metabolism of HDL components
[66]. For example, nephrotoxic syndrome induces a significant reduced expression of hepatic lipase in liver parenchyma, causing decreasing uptake of HDL triglycerides and phospholipids, increasing circulating cholesterol droplets, and impairing HDL maturation
[67]. Therefore, augmented renal proteinuria caused profound alteration in plasma and liver enzymes function, avoiding HDL maturation, determining profound impact in several organs including kidneys
[67].
It is clear that kidney injury could alter HDL levels and composition, but further studies are needed to evaluate the effects of other receptors, as SR-BI and ABACG1 in renal HDL catabolism or transcytosis.
4. SARS-CoV-2 Exploits the SR-BI Associated HDL to Promote its Entry
Despite the great efforts to elucidate the molecular mechanisms of SARS-CoV2 cell entry, the pathogenetic mechanisms have not been fully clarified and there are no effective therapies to stop or reverse the occurred infection. The partial inhibition obtained by ACE-2 antibodies in contraposition to maximum neutralization efficacy of monoclonal antibodies targeting the SARS-2-S1 N-terminal domain suggested that viral particles exploited additional receptors and channels for host cell entry
[68][69][70]. In accordance, besides ACE-2, other receptors, such as Dipeptidyl peptidase 4 (DPP4) or aminopeptidase N, have been proposed to play a role in SARS-CoV2 entry in target cells. Specifically, DPP4, also known as cluster of differentiation 26 (CD26), a serine exopeptidase expressed ubiquitously in lung, kidney, liver, gut, and immune cells, has been investigated to allow virus SARS-CoV-2 cell adhesion/virulence
[71].
Among additional mechanisms of enhancement SARS-CoV-2 entry, SR-BI has been recently examined to play a central role in augmenting virus attachment. As previously underlined, SR-BI is a kind of membrane protein with a molecular weight of 82 kDa, that acts as the major HDL scavenger receptor and mediates the selective HDL-cholesterol uptake of cells
[72]. This cholesterol delivery system is well studied in hepatocytes as well as steroidogenic cells as adipocytes, adrenal cells but also in fibroblasts, macrophages, ovarian cells, and testicular Leydig cells
[73]. Notably, epithelial alveolar type II cells also express SR-BI, in this location, SR-BI is involved in vitamin E intake, specially from HDL
[74][75].
In an interesting in vitro study, Wei et al. demonstrated that SARS-COV2 exploited the physiological function of SR-BI to mediate cholesterol-bound HDL intake to promote its cellular entry
[70]. In other words, even if SR-BI cannot bind to the SARS-2-S protein directly, the receptor acts as a linker molecule that recruits viral particles to come into proximity with ACE-2 by association with HDL. In accordance, the SR-BI expression conferred the greatest cell susceptibility to SARS-CoV-2 only when co-expressed with ACE-2. Therefore, it emerged that treatment of cultured cells with pharmacological SR-BI antagonists, could inhibit HDL-enhanced SARS-CoV-2 infection inhibition. Furthermore, also blockade of the cholesterol-binding site on SARS-2-S1 with a monoclonal antibody showed similar effects. In line, ITX 5601, a clinically approved inhibitor of HCV infection, strongly inhibits SARS-CoV-2 infection of cultured cells. Strikingly, besides lung, authors further showed that SR-BI is co-expressed with ACE2, predominately in lower respiratory tissues that are more affected by COVID-19 and in several extrapulmonary tissues as retina, testis, as well as kidney which could indicate an extended trophism for extrapulmonary tissues, thereby to the multiple-organ pathologies of COVID-19
[76].
SARS-COV-2 has a lipid envelope that merges with the host cell through endocytosis, internalizing its components in the cell. Recently, cell experiments showed that the susceptibility of the virus to fusion with the host cell membrane augmented when cholesterol was added to medium culture
[77]. For example, cholesterol-supplemented mouse fibroblasts showed increased susceptibility to fusion with murine hepatitis virus
[78]. ACE-2 resides mainly within lipid rafts, furthermore, in lipid raft areas, there are also caveolins, clathrins, and dynamin, that are molecules with a central role in the incorporation of viruses
[79]. Similarly, uptake mechanisms dependent from these molecules and on the presence of lipid rafts have been also identified for the simian virus (SV40)
[77].
In the context of HDL and lipoproteins metabolism, it could be interesting to note the existence of an association between ACE insertion/deletion polymorphism with HDL level and cardiovascular disease risk factors. As recently demonstrated, an ACE polymorphism in men was significantly associated with reduced HDL cholesterol levels
[80]. Cholesterol is an essential lipid component of vertebrate cell membranes, mainly through lipid rafts, i.e., microdomains enriched in cholesterol and sphingolipids. Lipid rafts have been suggested to play a fundamental role in several biological processes such as signal transduction, membrane trafficking, cytoskeletal organization
[81][82].
Given their unique protein composition, lipid raft microdomains are essential for the endocytosis-mediated process and serve as a platform and docking site for viruses to enter the host cell
[79][81]. Emerging evidence revealed that cholesterol diminution from cellular membranes has been shown to hamper SARS-CoV-2 infection
[83]. This finding led several researchers to speculate that SARS-CoV-2 may exploit the physiological function of SR-BI to achieve its entry and fusion processes
[84].
Furthermore, Meher et al. reported the effect of membrane cholesterol on the structure of the fusion peptide (residues 770–788) of S2 glycoprotein of SARS-CoV. Authors elegantly demonstrated that S2 binding affinity augmented with increasing levels of membrane cholesterol
[85]. On the other hand, cholesterol decline disturbs the virion membrane. Through the interference with lipid-dependent attachment to human host cells, sterols and cyclodextins can reduce the infectivity of CoVs.
[86]. In vitro depletion of membrane-bound cholesterol in ACE2-expressing cells led to a reduced infectivity of CoVs, since the binding of the spike protein was reduced by half. The mechanism of employing lipid raft rich in cholesterol together with the utilization of another type of lipid called monosialotetrahexosylganglioside 1 (GM1), to enter mammalian cells in culture is shared by both SARS-CoV and SARS-CoV-2. This concept has emerged by the reduction of infection in cells treated with a compound called methyl-β-cyclodextrin (MβCD) able to deplete cholesterol
[87]. The cholesterol removal by MβCD significantly dissociate the number of bonds between ACE-2 membrane protein and viral S glycoproteins
[88].
Some studies showed that MβCD treatment dose-dependently reduced expression of ACE-2 in the cell membrane, also reducing the infectivity of SARS-CoV2
[78]. The lipophilic core permits the contact of these molecules with lipid rafts. These harmless macromolecules are able to mimic binding domains for the enveloped virus, competing with host cell attack sites, thereby reducing infection.
Additionally, interaction of phytosterols with lipid raft molecules can lead to a decrease of membrane cholesterol content or destabilization of its structure, thereby affecting viral infectivity
[78]. In addition, the viral infectivity is modulated by homeostatic control of cholesterol content and fatty acid metabolism
[89]. More recently, Henrich, S. E. et al. elegantly demonstrated that SARS-CoV-2 viral entry is impaired by SR-BI genetic knockdown, suggesting that SR-BI is a co-receptor for SARS-CoV-2. Interestingly, authors also demonstrated that inorganic core nanoparticles around which HDL-associated protein (apolipoprotein A-I) and lipids were clustered (HDL-particles) targeted SR-BI to inhibit SARS-CoV-2 entry. Indeed, the superficial resemblance to HDLs caused these nanoparticles to firmly bind to SR-BI. These nanoparticles are characterized by the ability to strongly inhibit the entry of exosomes, which are extracellular lipid vesicles. Finally, by altering the type of lipids assembled on the surface of these nanoparticles, they could target Gram-negative bacterial LPS and prevent the LPS-mediated release of potent pro-inflammatory signalling.
Based on these findings, HDL nanoparticles could offer a promising strategy to prevent infection with SARS-CoV-2. Strikingly, they could also be investigated as a possible treatment for other cholesterol-dependent viral infections that are also based on lipid rafts for their successful entry into host cells
[87].
 +1 point
+1 point