 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kavita Y. Sarin | + 1478 word(s) | 1478 | 2021-08-03 09:38:22 | | | |
| 2 | Enzi Gong | Meta information modification | 1478 | 2021-08-04 02:39:23 | | |
Video Upload Options
Basal cell carcinoma (BCC) is a significant public health concern, with more than 3 million cases occurring each year in the United States, and with an increasing incidence. The molecular basis of BCC is complex, involving an interplay of inherited genetic susceptibility, including single nucleotide polymorphisms and genetic syndromes, and sporadic somatic mutations, often induced by carcinogenic exposure to UV radiation.
1. Overview
Basal cell carcinoma is the most common human cancer worldwide. The molecular basis of BCC involves an interplay of inherited genetic susceptibility and somatic mutations, commonly induced by exposure to UV radiation. In this review, we outline the currently known germline and somatic mutations implicated in the pathogenesis of BCC with particular attention paid toward affected molecular pathways. We also discuss polymorphisms and associated phenotypic traits in addition to active areas of BCC research. We finally provide a brief overview of existing non-surgical treatments and emerging targeted therapeutics for BCC such as Hedgehog pathway inhibitors, immune modulators, and histone deacetylase inhibitors.
Basal cell carcinoma (BCC) is a significant public health concern, with more than 3 million cases occurring each year in the United States, and with an increasing incidence. The molecular basis of BCC is complex, involving an interplay of inherited genetic susceptibility, including single nucleotide polymorphisms and genetic syndromes, and sporadic somatic mutations, often induced by carcinogenic exposure to UV radiation. This review outlines the currently known germline and somatic mutations implicated in the pathogenesis of BCC, including the key molecular pathways affected by these mutations, which drive oncogenesis. With advances in next generation sequencing and our understanding of the molecular genetics of BCC, established and emerging targeted therapeutics are offering new avenues for the non-surgical treatment of BCC. These agents, including Hedgehog pathway inhibitors, immune modulators, and histone deacetylase inhibitors, will also be discussed.
2. Basal Cell Carcinoma
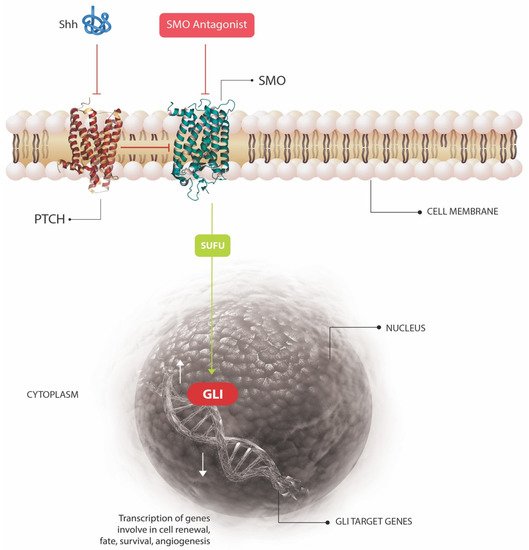
Of all human cancers, basal cell carcinoma (BCC) is the most common worldwide, and in many countries, its incidence continues to increase, representing a significant public health burden [1]. Each year in the United States, more than 3 million cases occur, and one in five individuals in the U.S. are estimated to develop at least one BCC during their lifetime [2]. In addition to the potential morbidity of BCC, the cancer has a significant economic impact, with annual U.S. healthcare expenditure for these tumors reaching almost $4 billion [3][4][5]. The molecular pathogenesis of BCC is complex, and involves an interplay of inherited genetic susceptibility [6] and sporadic somatic mutations [7]. The former predisposes an individual towards the development of BCC, and can include single nucleotide polymorphisms (SNPs), inherited disorders, and genetic traits [6], but the latter is generally required to induce carcinogenesis. While the types of variants that induce susceptibility to BCC are varied, the sporadic mutations often function to activate the Hedgehog (HH) signaling pathway, a growth and development pathway integral to the pathogenesis of BCC. This review will discuss in more depth the currently known germline and somatic mutations implicated in the pathogenesis of BCC, as well as the underlying molecular pathways affected. We will also give an overview of established and emerging targeted, non-surgical, therapeutics which could revolutionize the treatment of this common and important skin cancer.
3. Genetic Syndromes Associated with BCC Development
References
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A Systematic Review of Worldwide Incidence of Nonmelanoma Skin Cancer. Br. J. Dermatol. 2012, 166, 1069–1080.
- Miller, D.L.; Weinstock, M.A. Nonmelanoma Skin Cancer in the United States: Incidence. J. Am. Acad. Dermatol. 1994, 30, 774–778.
- Epstein, E.H. Basal Cell Carcinomas: Attack of the Hedgehog. Nat. Rev. Cancer 2008, 8, 743–754.
- Guy, G.P.; Machlin, S.R.; Ekwueme, D.U.; Yabroff, K.R. Prevalence and Costs of Skin Cancer Treatment in the U.S., 2002−2006 and 2007−2011. Am. J. Prev. Med. 2015, 48, 183–187.
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the US Population, 2012. JAMA Derm. 2015, 151, 1081–1086.
- Choquet, H.; Ashrafzadeh, S.; Kim, Y.; Asgari, M.M.; Jorgenson, E. Genetic and Environmental Factors Underlying Keratinocyte Carcinoma Risk. JCI Insight 2020, 5, 134783.
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485.
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth Incidence and Prevalence of Tumor-Prone Syndromes: Estimates from a UK Family Genetic Register Service. Am. J. Med. Genet. A 2010, 152, 327–332.
- Anderson, D.E.; Taylor, W.B.; Falls, H.F.; Davidson, R.T. The Nevoid Basal Cell Carcinoma Syndrome. Am. J. Hum. Genet. 1967, 19, 12–22.
- Jones, E.A.; Sajid, M.I.; Shenton, A.; Evans, D.G. Basal Cell Carcinomas in Gorlin Syndrome: A Review of 202 Patients. J. Skin Cancer 2011, 2011, 217378.
- Yasar, B.; Byers, H.J.; Smith, M.J.; Lear, J.; Oudit, D.; Bholah, Z.; Roberts, S.A.; Newman, W.G.; Evans, D.G. Common Variants Modify the Age of Onset for Basal Cell Carcinomas in Gorlin Syndrome. Eur. J. Hum. Genet. 2015, 23, 708–710.
- Kimonis, V.E.; Goldstein, A.M.; Pastakia, B.; Yang, M.L.; Kase, R.; DiGiovanna, J.J.; Bale, A.E.; Bale, S.J. Clinical Manifestations in 105 Persons with Nevoid Basal Cell Carcinoma Syndrome. Am. J. Med. Genet. 1997, 69, 299–308.
- Evans, D.G.; Oudit, D.; Smith, M.J.; Rutkowski, D.; Allan, E.; Newman, W.G.; Lear, J.T. First Evidence of Genotype-Phenotype Correlations in Gorlin Syndrome. J. Med. Genet. 2017, 54, 530–536.
- Farndon, P.A.; Del Mastro, R.G.; Evans, D.G.; Kilpatrick, M.W. Location of Gene for Gorlin Syndrome. Lancet 1992, 339, 581–582.
- Lam, C.-W.; Leung, C.-Y.; Lee, K.-C.; Xie, J.; Lo, F.-M.; Au, T.-S.; Tong, S.-F.; Poon, M.-K.; Chan, L.-Y.; Luk, N.-M. Novel Mutations in the PATCHED Gene in Basal Cell Nevus Syndrome. Mol. Genet. Metab. 2002, 76, 57–61.
- Lo Muzio, L. Nevoid Basal Cell Carcinoma Syndrome (Gorlin Syndrome). Orphanet J. Rare Dis. 2008, 3, 32.
- i Altaba, A.R.; Sánchez, P.; Dahmane, N. Gli and Hedgehog in Cancer: Tumours, Embryos and Stem Cells. Nat. Rev. Cancer 2002, 2, 361–372.
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; i Altaba, A.R. Activation of the Transcription Factor Gli1 and the Sonic Hedgehog Signalling Pathway in Skin Tumours. Nature 1997, 389, 876–881.
- Booth, D.R. The Hedgehog Signalling Pathway and Its Role in Basal Cell Carcinoma. Cancer Metastasis Rev. 1999, 18, 261–284.
- Torrelo, A.; Sprecher, E.; Mediero, I.G.; Bergman, R.; Zambrano, A. What Syndrome Is This? Bazex-Dupre-Christol Syndrome. Pediatr. Dermatol. 2006, 23, 286–290.
- Bal, E.; Park, H.-S.; Belaid-Choucair, Z.; Kayserili, H.; Naville, M.; Madrange, M.; Chiticariu, E.; Hadj-Rabia, S.; Cagnard, N.; Kuonen, F.; et al. Mutations in ACTRT1 and Its Enhancer RNA Elements Lead to Aberrant Activation of Hedgehog Signaling in Inherited and Sporadic Basal Cell Carcinomas. Nat. Med. 2017, 23, 1226–1233.
- DiGiovanna, J.J.; Kraemer, K.H. Shining a Light on Xeroderma Pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796.
- Black, J.O. Xeroderma Pigmentosum. Head Neck Pathol. 2016, 10, 139–144.


