

microRNAs represent the most studied type of small ncRNAs and it has been demonstrated that miRNAs play essential roles in multiple biological contexts, including normal development and diseases. Cardiac arrhythmias are prevalent among humans across all age ranges, affecting millions of people worldwide. While cardiac arrhythmias vary widely in their clinical presentation, they possess shared complex electrophysiologic properties at cellular level that have not been fully studied.
- cardiac arrhythmia
- microRNAs
- lncRNAs
- cardiac action potential
1. The Electrical Components of the Adult Heart
Rhythmic contraction of the heart, leading to alternative systole and diastole contraction phases is controlled by the cardiac conduction system (CCS). The CCS is formed by slow and fast conduction pathways. The slow components are two distinct low conducting and self-firing nodes, the sinoatrial and the atrioventricular node, respectively. The sinoatrial node is located at the junction between the right superior caval vein entrance and the atrial chamber myocardium and is the main pacemaker of the heart [2]. The atrioventricular node is located at the top of the interventricular septum just at the junction between atrial and ventricular myocardium. The fast conducting components of the cardiac conduction system are exclusively located in the ventricular chambers, and are composed by the bundle of His, the left and right bundle branches, and the Purkinje fiber network [2].
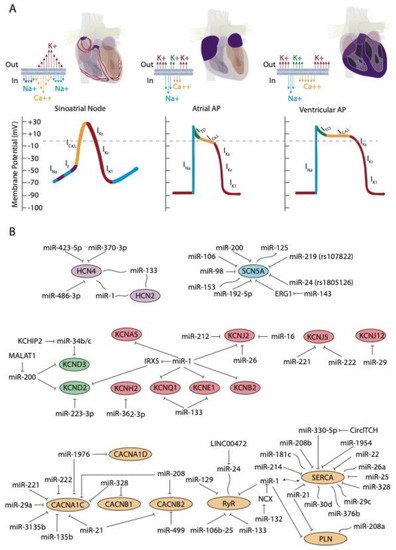
At cellular level, the electrical activity of the myocardial cells is governed by an exquisite balance of inward and outward ion currents that configure the cardiac action potential. The cardiac action potential can be divided in at least four different phases. The first phase is initiated with a rapid upstroke of inward sodium currents, leading to the depolarization phase. Subsequently, the repolarization phase is initiated with fine-tuned balance of outward potassium currents, leading to phases two (ITO currents) and three (IK currents) of the cardiac action potential to finally reach the fourth phase of resting membrane potential (IK1 currents) [3,4,5]. During the plateau phase, calcium contraction coupling also takes place, by the activation of the ICa,L currents followed by mobilization of intracellular calcium from the sarcoplasmic reticulum, throughout the calcium-induced calcium release mechanism [6,7,8]. This general configuration of the cardiac action potential, although applicable to all cardiomyocytes, displays subtle variations on distinct cardiac regions. Importantly, such variability is due to distinct molecular substrates governing such events, such as for example, the upstroke phase in the cardiac action potential of the cardiac conduction system is governed by cation channels, with limited contribution of the sodium channels [4,5]. In addition to the distinct functional properties of the cardiac conduction system, the working myocardium also display significant differences between each cardiac regions—e.g., between atrial and ventricular myocardium—and also within the ventricular myocardium itself—e.g., epicardial vs. endocardial configurations. Such regional differences are mainly motivated by regional differences in the relative contribution of the outward potassium channels governing the rapid (ITO; IKur), plateau (IKr; IKs), and final (IKs; IK1) phases of the cardiac action potential repolarization as well as by the L-type calcium channels in the plateau phase. Finally, it is important to highlight that there are notable species-specific differences in the contribution of the discrete ion currents to the final configuration of the cardiac action potential. Such differences are particularly applicable to the repolarization phase in distinct experimental models such as rat, mouse, pig, and zebrafish as compare to humans, as widely documented elsewhere [9,10,11,12,13,14,15].
2. Role of ncRNAs in the Cardiac Action Potential
2.1. ncRNAs in the Upstroke Phase (INa Currents)
The upstroke phase of the cardiac action potential in fast conducting cells, i.e., atrial and ventricular myocytes, is primarily modulated by the fast INa current (NaV1.5) with a smaller contribution of Nav1.8. Importantly, the function of the pore-forming Nav1.5 channel is also modulated by ancillary subunits such as Navβ1-Navβ4 (Figure 1A). SCN5A encodes the voltage-gated Na+ channel NaV1.5. Mutations in SCN5A are associated to inherited arrhythmias and cardiomyopathy [35,36,37,38]. Moreover, single-nucleotide polymorphisms (SNPs) linked to SCN5A splicing, localization, and function are also associated to sudden cardiac death [39,40]. SCN10A encodes the voltage-gated Na+ channel NaV1.8. Importantly, mutations in SCN10A have also been linked to sudden unexplained death [41], atrial fibrillation [42,43], and Brugada syndrome [40,44,45]. Furthermore, SCN5A and SCN10A share common regulatory elements that are relevant for cardiac function [46].
| Current | microRNA | Gene | Function | Reference |
|---|---|---|---|---|
| INa | miR-98, miR-106, miR-200, miR-219, miR-125, miR-153 | SCN5A | INa ↑/INa ↓ | [47,53] |
| miR-192-5p | INa ↓ | [48] | ||
| miR-200c | - | [50] | ||
| miR-143 | INa ↓ | [51] | ||
| miR-24 | INa ↓ | [52] | ||
| If | miR-423-5p | HCN4 | If ↓ | [63] |
| miR-370-3p | If ↓ | [64] | ||
| miR-486-3p | If ↓ | [65] | ||
| miR-1, miR-133 | If ↑ | [66,67,68,69] | ||
| miR-1, miR-133 | HCN2 | If ↑ | [66,67,68,69] | |
| ITO | miR-1 | KCND2 | ITO ↓ | [70] |
| miR-223-3p | ITO ↓ | [71] | ||
| miR-34b/c | ITO = | [72] | ||
| miR-200 | ITO ↓ | [73] | ||
| miR-200 | KCND3 | ITO ↓ | [73] | |
| IKur | miR-1 | KCNA5 | IKur ? | [74] |
| IKr | miR-134, miR-103a-1, miR-143, miR-3619 | hERG | IKr ↓ | [75] |
| IKS | miR-1, miR-133 | KCNE1 | IKS ↓ | [76,77] |
| miR-1, miR-133 | KCNQ1 | IKS ↓ | [76] | |
| miR-1, miR-133 | KCNB2 | IKS ↓ | [77] | |
| IK1 | miR-1 | KCNJ2 | IK1 ↓/IK1 ↑ | [49,78,79] |
| miR-16 | IK1 ↓ | [80] | ||
| miR-26 | IK1 ↑ | [81,82] | ||
| miR-212 | IK1 ↓ | [83,84] | ||
| miR-29 | KCNJ12 | IK1 ↓ | [85] | |
| miR-221/222 | KCNJ5 | IK1 ↓ | [86] | |
| ICa,L | miR-328 | CACNA1C | ICa,L ↓ | [87] |
| miR-21, miR-208b | ICa,L ↓ | [88,89] | ||
| miR-20a, miR-3135b | ICa,L ↓ | [90] | ||
| miR-499 | ICa,L ↓ | [91] | ||
| miR-135b | ICa,L ↓ | [92] | ||
| miR-221/222 | ICa,L ↓ | [86] | ||
| miR-328 | CACNB1 | ICa,L ↓ | [87] | |
| miR-21, miR-208b | CACNB2 | ICa,L ↓ | [88,89] | |
| miR-499 | ICa,L ↓ | [91] | ||
| miR-329 | ICa,L ↓ | [91] | ||
| CICR | miR-106b | RYR2 | - | [93,94] |
| miR-129 | [95] | |||
| miR-1, miR-133 | [96,97] | |||
| miR-23 | [98,99] | |||
| miR-25 | SERCA2A | - | [100,101] | |
| miR-328 | [102,103] | |||
| miR-29c | [104] | |||
| miR-21 | [105] | |||
| miR-208b | [89] | |||
| miR-22 | [106] | |||
| miR-214 | [107] | |||
| miR-1954 | [108] | |||
| miR-376b, miR-1, miR-26a, miR-30d, miR-181 | [109] | |||
| miR-1, miR-21 | PLN | - | [110] | |
| miR-208a | [111] | |||
| miR-132 | NCX1 | - | [112] | |
| miR-1 | [107] |
| Gene | Disease | Alteration | Mir Related | Reference |
|---|---|---|---|---|
| SCN5A | Inherited arrhythmias and cardiomyopathy | Mutation | - | [35,36,37,38] |
| Sudden death | SNPs | - | [39,40] | |
| Brugada syndrome | SNPs/↓ expression | miR-219 | [47,53] | |
| Atrial fibrillation | ↓ expression | miR-192-5p | [48] | |
| Heart failure | SNPs/↓ expression | miR-24 | [52] | |
| SCN10A | Sudden death | Mutation | - | [41] |
| Atrial fibrillation | - | [42,43] | ||
| Brugada syndrome | - | [29,44,45] | ||
| HCN4 | Bradycardia | ↓ expression | miR-423-5p, miR-370-3p | [63,64] |
| Age atrial fibrillation | ↑ expression | miR-1, miR-133 | [66] | |
| Myocarial infarction | ↑ expression | miR-1, miR-133 | [67,68] | |
| HCN2 | Age atrial fibrillation | ↑ expression | miR-1, miR-133 | [66] |
| Myocardial infarction | ↑ expression | miR-1, miR-133 | [67,68] | |
| KCND2 | Sudden death | ↓ expression | miR-1 | [70] |
| Acute myocardial infarction | ↓ expression | miR-223-3p | [71] | |
| Myocardial infarction | ↓ expression | miR-200c | [73] | |
| KCNH2 | LQT syndrome (type 2) | Mutation | - | [113] |
| Heart failure | ↓ expression | miR362-3p | [114] | |
| KCNE2 | ||||
| LQT syndrome (type 6) | ||||
| LQT syndrome (type 1) | LQT syndrome (type 1) | ↓ expression | - | [115,116,117] |
| Atrial fibrillation | ↓ expression | miR-1 | [77] | |
| KCNB2 | ||||
| Atrial fibrillation | Myocardial infarction | ↓/↑ expression | miR-1, miR-16 | [78,81] |
| Atrial fibrillation | ↑ expression | miR-1, miR-26 | [49,79,82] | |
| Heart failure | ↓ expression | miR-212 | [84] | |
| KCNJ12 | Myocardial infarction | ↓ expression | miR-29 | [85] |
| KCNJ5 | Atrial fibrillation | ↓ expression | miR-221/222 | [86] |
| CACNA1C | Atrial fibrillation | ↓ expression | miR-221/222 | [86] |
| miR-328 | [87] | |||
| miR-21 | [88] | |||
| miR-208b | [89] | |||
| miR-29b, miR-3135b | [90] | |||
| CACNB2 | Atrial fibrillation | ↓ expression | miR-21 | [88] |
| miR-208b | [89] | |||
| miR-499, miR-329 | [91] | |||
| RYR2 | Atrial fibrillation | ↑ expression | miR-106b-25 | [93,94] |
| miR-106a, miR-93 | [94,118] | |||
| miR-129* | [95] | |||
| miR-1*, miR133* | [96,97] | |||
| miR-24* | [98,99] | |||
| SERCA2A | Atrial fibrillation | ↓ expression | miR-25 | [100,101] |
| miR-328 | [102,103] | |||
| miR-29c | [104] | |||
| miR-21*, miR-208b*, | [105,89] | |||
| miR-214*, miR-1954*, | [107,108] | |||
| miR-376b, miR-1*, | [109] | |||
| miR-26a*, miR-30d*, | [109] | |||
| miR-181c* | [109] | |||
| miR-330-5p* | [119] | |||
| PLN | Cardiac arrhythmias | - | miR-1, miR-21 | [98] |
| miR-208a* | [99] | |||
| NCX1 | Cardiac arrhythmias | - | miR-132 | [112] |
| miR-1 | [107] |
3.2. ncRNAs in the Upstroke Phase (If Current)
The hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are the structural pore-forming subunits governing this current, with four HCN isoforms known (HCN1-4), among which HCN4 is the most highly expressed in the sinoatrial and atrioventricular nodes (Figure 1A).
D’Souza et al. [63] reported a direct biochemical interaction between miR-423-5p and HCN4 and they further demonstrated that miR-423-5p contributes to training-induced bradycardia by targeting HCN4. Thus miR-423-5p modulates the If current and the heart rate in mice. Yanni et al. [64] reported direct interaction between miR-370-3p and HCN4 (Figure 1B).
Indirect evidence on the role of miR-1 and miR-133 regulating HCN isoforms have been also reported. Inversed expression patterns of HCN2 and HCN4 (upregulated) and miR-1 and miR-133 (downregulated) have been reported in age-associated atrial fibrillation [66] myocardial infarction (MI) [67,68], and exercise training [69], yet it remains to be established if these microRNAs can direct target HCN isoforms (Figure 1B).
Similarly, no evidence has been reported to date on the direct functional role of microRNAs regulating HCN4 in arrhythmogenic syndromes, supporting the notion that additional studies are required in this context. A summary of the microRNA interaction with the HCN channels is provided on Table 1, while their links to distinct cardiac diseases is provided on Table 2.
3.3. ncRNAs in Sodium Channel Interacting Proteins
Calmodulin has been extensively reported to directly interact with NaV1.5 (SCN5A) sodium channel and thus to modulate its function [128,129,130,131,132,133,134,135]. Although distinct microRNAs such as miR-1 [136], let-7a [137], miR-625-5p [138], miR-525-5p [139], miR-338-5p [140], miR-185 [141], miR-145 [142], miR-30b-5p [143], and miR-675 [144] have been reported to modulate calmodulin expression, these reports exclusively describe their functional role in cardiac hypertrophy and failed to provide a direct link to sodium channel regulation. To date, the only report linking microRNAs, i.e., miR-26a, and cardiac arrhythmias, i.e., atrial fibrillation, is reported by Qi et al. [145]. Thus, the plausible contribution to sodium channel function by calmodulin interactive protein remains elusive.
4. Role of ncRNAs in Cardiac Repolarization
4.1. ncRNAs in the Early Repolarization (ITO Transient Outward K+ Current)
After cardiac depolarization, the early repolarization process is governed by cardiac transient outward potassium current (ITO). ITO is rapidly activated after a fast increase of the membrane potential, where a short-lived, hyperpolarizing outward K+ current (ITO) makes K+ ions from inside the cells to flow to the extracellular space, causing the transmembrane voltage to decrease. ITO is then quickly deactivated, stopping the repolarization and ending the first phase of the action potential [155].
Several microRNAs have been described to be involved in the regulation of these channels. In particular, in 2007, Zhao and co-workers [70], demonstrated that Kcnd2 is positively regulated by miR-1, through Irx5 inhibition in mice, thus altering the endocardial to epicardial transmural gradient controlled by Kcnd2 within the ventricular cardiomyocytes and thus resulting in ventricular repolarization abnormalities. Kcnd2 is also regulated by miR-223-3p, a microRNA that is remarkably upregulated in a rat model of acute MI and consequently, Kv4.2 protein levels and ITO density were significantly decreased [71] (Figure 1B). Such impaired modulation of Kv4.2 protein expression and thus of ITO current can cause prolongation of the action potential duration and thus promote arrhythmias.
4.2. ncRNAs in the Plateau Phase and Terminal Repolarization (IKr, IKs, IKur K+ Current)
4.3. ncRNAs Modulating the Ultra-Rapid Delayed Rectifier K+ Current (IKur)
4.4. ncRNAs Modulating the Rapid Delayed Rectifier K+ Current (IKr)
IKr currents are governed by hERG channels, also known as Kv11.1 [161]. As an homolog of the Drosophila “ether-a-go-go” (EAG) potassium channel, hERG was first cloned in the brain [162]. hERG channels are encoded by KCNH2 and mutations in KCNH2 have been associated to long QT syndrome (type 2; LQTS2) [113]. Ancillary MiRP1 (or KCNE2) subunits, that constitute single transmembrane protein homologous to KCNE1, was shown to associate with HERG channels and modulate IKr biophysical properties [163]. Mutations in KCNE2 have also been associated to long QT syndrome (type 6; LQTS6) [163].
4.5. ncRNAs Modulating the Slow Delayed Rectifier K + Current (IKs)
To date, scarce evidence is available regarding the functional impact of ncRNAs in IKs current modulation. Li et al. [76] examine miR-1/miR-133 levels, the potassium channel KCNE1 and KCNQ1 levels and IKs current in cardiac progenitor cells (CPCs) of normal human hearts. These authors observed that human CPCs expressed KCNE1 and KCNQ1 and possessed functional IKs currents (Figure 1B).
4.6. ncRNAs in the Resting Membrane Potential (IK1 Current and Na,K ATPase)
Several microRNAs have been reported to modulate IK1 current in distinct biological contexts. miR-1 levels are increased in patients with coronary artery diseases (CAD) and also in an experimental rat model of MI. In this context, miR-1 silences KCNJ2 protein expression, and also GJA1, by directly targeting their 3´UTRs, respectively [78] (Figure 1B). On the contrary hand, miR-1 levels are greatly reduced in human AF as well as in AF experimental models, contributing to upregulation of Kir2.1 subunit, leading thus to increased IK1, being this upregulation of inward-rectifier currents important for AF maintenance [49,79]. Additionally, miR-16 overexpression suppress KCNJ2/Kir2.1 expression in a rat experimental model of MI [80].
A summary of the microRNA interaction with the potassium channels is provided on Table 1, while their links to distinct cardiac diseases is provided on Table 2.
5. Role of ncRNAs in Conduction Contraction Coupling
5.1. ncRNAs in Calcium Currents (ICa,L Current)
Transgenic mice overexpressing miR-1 resulted in severe electrophysiological defects, causing atrioventricular block [184]. Molecular analysis demonstrates that several key components contributing to the electrical wiring of the heart were altered, such as Cx43 and Kir2.1. Electrophysiological studies revealed that ICa and IK1 currents were decreased. Knockdown of miR-1 overexpression using LNA-anti-miR-1 administration reversed such electrophysiological alteration, demonstrating a pivotal role for miR-1 in cardiac electrophysiology and particularly in calcium homeostasis. More recently, Zhang et al. (2019) demonstrated that transgenic mice overexpressing miR-1976 directly targeted two key calcium channels (i.e., Cav1.2 and Cav1.3, encoded by CACNA1C and CACNA1D, respectively) resulting in SAN dysfunction and thus lower heart rates, a phenotype reminiscent of sick sinus node syndrome in humans [185].
5.2. The Role of ncRNAs in Calcium-Induced Calcium Release
6. Conclusions and Perspectives
This entry is adapted from the peer-reviewed paper 10.3390/hearts2030026
