Apoptosis is an evolutionarily conserved and tightly regulated cell death pathway. Physiological cell death is important for maintaining homeostasis and optimal biological conditions by continuous elimination of undesired or superfluous cells. The BH3-only pro-apoptotic members are strong inducers of apoptosis. The pro-apoptotic BH3-only protein Noxa activates multiple death pathways by inhibiting the anti-apoptotic Bcl-2 family protein, Mcl-1, and other protein members leading to Bax and Bak activation and MOMP. On the other hand, Puma is induced by p53-dependent and p53-independent apoptotic stimuli in several cancer cell lines. Moreover, this protein is involved in several physiological and pathological processes, such as immunity, cancer, and neurodegenerative diseases. Future heat shock research could disclose the effect of hyperthermia on both Noxa and BH3-only proteins. This suggests post-transcriptional mechanisms controlling the translation of both Puma and Noxa mRNA in heat-shocked cells.
1. Introduction
Apoptosis is a regulated form of genetically programmed cell death that has significant roles in development, tissue homeostasis, and in response to environmental stresses. Regulated cell death was first described as shrinkage necrosis, due to morphological observations, and later renamed apoptosis. The term apoptosis is derived from the Greek prefix “apo-”, which means “to separate”, and the suffix “-ptosis”, which means ‘to fall off’ [
1,
2,
3]. During the first phase of apoptosis, termed the condensation phase, the cell separates from neighboring cells, the cytoplasm and nucleus become condensed, and nuclear DNA is fragmented [
3]. The endoplasmic reticulum (ER), Golgi apparatus and mitochondria become disrupted [
4]. This will intrigue the occurrence of regulated proteolysis [
5,
6]. The cell is then fragmented into apoptotic bodies [
3,
4]. The second stage of apoptosis involves phagocytosis and the destruction of apoptotic bodies [
3,
7]. Mutations in genes regulating apoptosis have been implicated in several human diseases including cancer [
8,
9,
10,
11,
12].
Autophagy and apoptosis are key biochemical mechanisms that keep organismal and cellular homeostasis in check. Autophagy preserves cellular homeostasis by recycling selected intracellular organelles and chemicals, whereas apoptosis fulfills its job by demolishing damaged or undesired cells. Autophagy is a cell’s process of degrading its own cytoplasmic material. In other words, the cell’s interior contents deteriorate. Autophagy is derived from the words “auto-” (self) and “phagy” (eating). As a result, autophagy causes the cell to practically consume itself. This digestion takes place in the lysosome, which is commonly referred to as a cell’s stomach [
13]. This mechanism, which permits intracellular organelles and proteins to be recycled, assists cells by eliminating damaged or undesirable organelles and macromolecules and providing energy and building materials for cellular operations and de novo biosynthesis [
14,
15].
The B-cell lymphoma-2 (Bcl-2) family proteins have a crucial role in controlling the apoptotic pathway [
19,
20,
21,
22,
23]. The pro-apoptotic homology (BH), BH3-only protein, and Phorbol-12-myristate-13-acetate-induced protein 1 (Noxa), activates multiple death pathways, which can be achieved by inhibiting the anti-apoptotic Bcl-2 family protein myeloid cell leukemia-1 (Mcl-1), and other protein members. This ultimately activates Bcl-2-associated X protein (Bax), Bcl-2 homologous antagonist killer (Bak), and major outer mitochondrial membrane (OMM) proteins [
19,
24,
25]. However, BH3-only protein, p53 upregulated modulator of apoptosis (Puma) is induced by p53-dependent and p53-independent apoptotic stimuli in several cancer cell lines. These family proteins are involved in many physiological and pathological processes [
26,
27], including the immune response [
28,
29,
30], cancer [
31,
32,
33,
34], and neurodegenerative diseases [
12,
35,
36,
37].
2. Bcl-2 Proteins and the Regulation of Mitochondrial Outer Membrane Permeability
Molecular insights into apoptosis first emerged during the 1980s and 1990s from a convergence of mammalian cancer cytogenetics and genetic studies on developmentally programmed cell death in
Caenorhabditis elegans [
38]. The previously unknown gene Bcl-2 was identified from the breakpoint region of a recurrent chromosomal translocation (18q21) in human follicular lymphoma [
7,
39]. The Bcl-2 family of proteins are central regulators of stress-induced apoptosis as they control diverse survival and death signals that are generated inside and outside the cell [
40,
41]. Bcl-2 proteins are found in all cells and consist of 12 distinct members, many of which are expressed as alternate isoforms. Structurally, they all contain at least one of the conserved sequence motifs called Bcl-2 BH domains. This family is functionally subdivided into two classes based on their activity and number of BH domains: anti-apoptotic and pro-apoptotic proteins including the BH3-only members [
19,
41,
42]. The mutual interaction between pro-apoptotic and anti-apoptotic members establishes the threshold that determines whether a cell should survive or die [
43]. In a sense, they act as checkpoints through which survival and death signals must pass to elicit the cell’s fate as they control stress-induced cell death [
44].
The anti-apoptotic Bcl-2 members, including Bcl-2 itself, Bcl-xL, Mcl-1, Bcl-W, and A1/BFL-1 share four conserved BH domains of structural homology. These proteins prevent cell death against diverse cytotoxic signals, both physiological and imposed. This maintains the integrity of the ER, mitochondrial, and nuclear membranes, thus protecting cells from apoptosis [
45,
46]. The proximity of BH 1, 2, and 3 form a hydrophobic pocket that operates as a receptor for the BH3 domain of the pro-apoptotic BH3-only members. The anti-apoptotic Bcl-2 proteins work together to inhibit the release of cytochrome c from the mitochondria by preventing the activation of the pro-apoptotic members Bax and Bak in the OMM [
44]. The anti-apoptotic proteins’ normal function is to prevent inappropriate cell death and therefore requires the neutralization of the pro-apoptotic proteins by the anti-apoptotic members [
47].
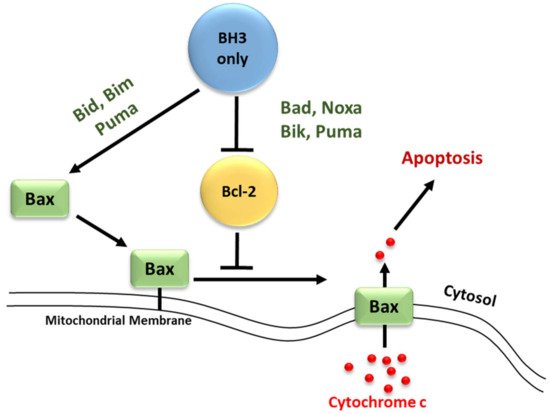
Just as the anti-apoptotic Bcl-2 proteins promote tumourigenesis when deregulated, the pro-apoptotic members function as tumor suppressors. The pro-apoptotic Bcl-2 members are divided into multi-domain effector proteins, such as Bax, Bak, and the less well-known Bok, as well as the large subgroup of BH3-only proteins, all of which trigger or sensitize the cell to apoptosis [
48,
49,
50]. The pro-apoptotic effector proteins Bax, Bak, and Bok possess three BH domains and adopt similar globular structures: a helical bundle surrounding a central hydrophobic core helix [
44,
51,
52]. This groove constitutes a crucial surface for interactions with the BH3 domain of pro-apoptotic members of the Bcl-2 family [
38]. These interactions primarily occur on intracellular membranes, such as that of the OMM, where many of the Bcl-2 family members are directed by their carboxy-terminal hydrophobic TM domain [
53]. Unlike the anti-apoptotic members, the active conformation of Bax/Bak damages rather than protects the OMM of stressed cells. This yields the formation of pores leading to membrane dysfunction [
54]. The most studied pro-apoptotic proteins Bax and Bak exist in an inactive monomeric state in healthy cells. In stressed cells, they are major inducers of mitochondrial outer membrane permeabilization (MOMP) [
55].
Recent studies have focused on the effects of the Bcl-2 family members on MOMP. Although over a decade has passed since the discovery of the BH3-only protein Puma, the question of how this protein activates Bax/Bak, consequently leading to MOMP, remains unresolved. At present, two mechanisms have been proposed concerning the relationship of the Bcl-2 protein family members employing direct or indirect activation of the mitochondrial apoptotic pathway, shown in
Figure 2 [
44,
47,
54]. Two models are explaining Bak/Bax activation through the interaction of the Bcl-2 protein family members: the direct activation model and the indirect activation model.
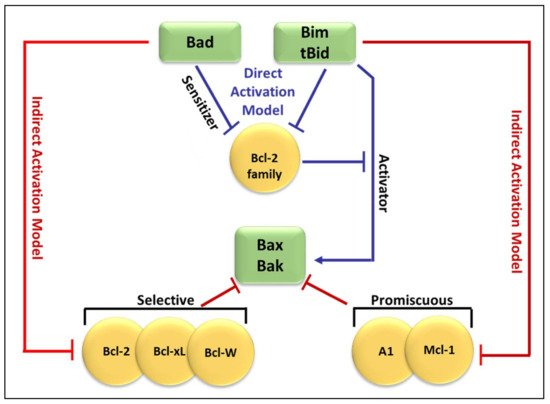
Figure 2. Direct and indirect activation of the intrinsic apoptotic pathway. Apoptosis is initiated by the release of cytochrome c from the mitochondria inter-membrane space to the cytosol via channels formed by the oligomerization of Bak or Bax in the OMM.
Figure 3 compares both the indirect and direct models of Bax and Bak activation [
47]. As shown in
Figure 3, in the direct model (blue arrows), Bim and tBid act as “activators” by binding to Bak and Bax directly to induce pore formation [
59]. The remaining BH3-only proteins act as “sensitizers” and bind to the anti-apoptotic Bcl-2-like proteins, releasing bound Bim and tBid and allowing them to directly activate Bak and Bax [
57,
66]. Additionally, some BH3-only proteins can only bind to specific anti-apoptotic Bcl-2 family proteins (selective) while others can bind to all anti-apoptotic Bcl-2 family proteins (promiscuous). This ability of BH3-only proteins to bind and neutralize certainly, or all, anti-apoptotic proteins is based on their BH3 domains [
67].
Figure 3. Comparison of the indirect and direct model of Bax and Bak activation. In the direct activation model (blue arrows), Bim and tBid act as “activators” by binding to Bak and Bax directly to induce pore formation. The remaining BH3-only proteins (Bid) bind to the Bcl-2-like proteins, releasing bound Bim and tBid which then bind directly to Bak and Bax. The indirect activation model (represented by red arrows from both sides), includes the engagement of anti-apoptotic Bcl-2-like proteins which then leads to the liberation of Bak and Bax.
3. Regulation of Apoptosis
3.1. Regulators of Cell Death: Caspases
Caspases (Cysteinyl aspartate-specific proteases) are a family of proteins activated by a variety of stimuli representing a vital step for the induction of apoptosis. These proteases initiate and control the cellular death pathway by cleaving a diverse set of cellular proteins [68].
3.2. Regulators of Cell Death: Bcl-2 Family Proteins
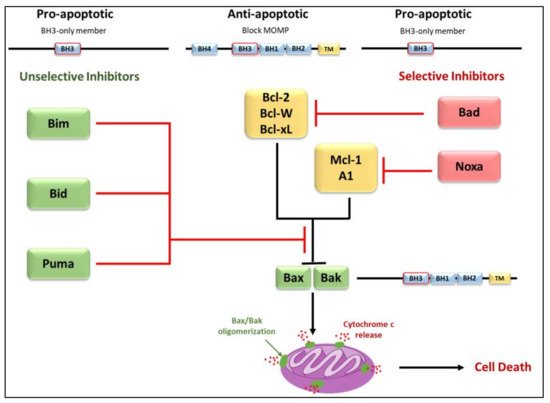
The convergence of pro-apoptotic signal-transducing molecules or cytotoxic stimuli to the outer mitochondrial membrane induces MOMP, releasing cytochrome c into the cytosol and promoting apoptosome formation and caspase activation (
Figure 4) [
18,
43,
72,
79,
80]. MOMP is under the control of the Bcl-2 family of proteins. They are subdivided into three categories; pro-apoptotic proteins containing three Bcl-2 Homology (BH) domains, pro-apoptotic BH3- only proteins lacking BH domains 1 and 2, and anti-apoptotic proteins [
1,
43,
72,
79,
81]. The anti-apoptotic Bcl-2 family proteins; Mcl-1, Bcl-2, Bcl-xL, Bcl-w, and A1, function to block MOMP. Overexpression of these proteins, which commonly occurs in cancer, inhibits stress-induced apoptosis [
18,
79]. The pro-apoptotic BH-1,2,3 proteins; Bax, Bak, and Bok, share three homology domains such as those of anti-apoptotic Bcl-2 family members and are considered essential for the progression of the cellular death pathway [
12,
79]. Finally, the pro-apoptotic BH3- only proteins; Bid, Bim, Bad, Noxa, and Puma do not have BH domains 1 and 2, possessing only the BH3 domain [
79,
81]. Interactions amongst the Bcl-2 proteins regulate MOMP [
1,
79,
81]. Suppression of Bax/Bak activation by the antiapoptotic Bcl-2 family members can be overcome by the pro-apoptotic BH3-only proteins (
Figure 4). These proteins act as stress sensors and are regulated both transcriptionally and through post-translational modifications [
47,
82].
Figure 4. Differential binding of BH3-only proteins in the inhibition or progression of MOMP. To alleviate the inhibition that anti-apoptotic members have on Bax/Bak oligomerization, either a single unselective BH3-only member or multiple selective BH3-only members must bind. Selective members have an affinity for specific anti-apoptotic members whereas unselective members may bind to all members. Unselective members Bim, Bid, and Puma bind with high affinity and are considered potent inducers of apoptosis. Due to the selective nature of Bad and Noxa, these members are considered weak inducers.
3.3. Elucidation of the Caspases Reaction Mechanism
Caspases stand for Cysteine (Cys)-dependent aspartate-directed proteases are a sub-branch of the enzymes family proteases which possess an indispensable role in apoptosis. The commonly proposed mechanism is divided into two principal parts. It describes the cleavage of the peptide bond (amide function).
For phase 1: Formation of the covalent adduct
-
Nucleophilic activation: the alkaline property of one of the nitrogen atoms, in the imidazolic part of Histidine (His), deprotonates the hydrogen in the thiol (-SH) group of the Cys residue, yielding a thiolate.
-
Thiolate nucleophilic attack on carbonyl: the carbonyl group of the aspartic peptide bond undergoes a nucleophilic attack by the yielded thiolate in the latter step. This contributes to the formation of a first tetrahedral intermediate.
-
α-amino protonation: the amine group of I1 constitutes a good leaving group. This will enhance the possibility of the protonation of the α-amino moiety by the previous protonated nitrogen of the His residue.
-
Formation of the covalent adduct: the acyl-enzyme complex and the cleavage of the peptide bond.
For phase 2: Hydrolysis of the covalent adduct
The catalytic cycle completion yields the formation of carboxylic acid from the starting peptide:
-
Once, again the alkaline property of one of the nitrogen atoms in the imidazolic part of His deprotonates a water molecule.
-
This deprotonation contributes to the formation of a hydroxide. The strong alkaline and nucleophilic property of the hydroxide contributes to the attack of the electrophilic site of the carbonyl function. This yields a second tetrahedral intermediate.
-
α-thio protonation: Similarly, to the third step of phase 1, the sulfur in I2 constitutes a good leaving group. This enhances the possibility of protonation of the α-thio moiety by the previously protonated nitrogen of the His residue.
-
Formation of the carboxylic acid by regeneration of His and Cys counterparts.
Sulpizi et al. [
88] proposed another mechanism and performed a Quantum Mechanics/Molecular Mechanics simulation to examine in detail the mechanism of caspase-3, focusing their attention on the hydrolysis of the covalent adduct phase (Phase 2). The proposed and simulated mechanism by these authors involves two water molecules, working as proton donors. The first water molecule worked on the formation of a hydrogen bond with the His. This interaction favors the alkaline/nucleophilic attack of water to the carbonyl function of Cys. The second one enters the active site from the solvent bulk during the molecular dynamic simulation. The results of this theoretical study, which discarded the previously postulated mechanism is summarized as follows:
-
The already formed hydrogen bond between His and the first water molecule will favor the deprotonation of the latter. The yielded hydroxide attacks the acyl-enzyme complex on the carbonyl moiety.
-
The yielded alkoxide in I3 will attack the proton already captured by the His part in (a). This will form a germinal diol (I4; Figure 6).
-
The carboxylate function of the side-chain aspartate will attack one of the hydrogens of the diol. The thiol, acting as a good leaving group, will enhance the possibility of the formation of a carbonyl bond; thereby a carboxylic acid and a thiolate (I5;
Figure 6). The computational investigation shows that for the attack of the water molecule, a free energy barrier of about 19 ± 4 kcal/mol must be overcome, these trends are following the experimental results of Sulpizi et al. [
88].
Following their calculations, Miscione et al. [
83] have revealed the existence of a novel mechanism that involves the activation of a catalytic dyad of His and Cys. The conventionally proposed path cannot take place because of the large distance between His’s nitrogen and Cys’s hydrogen. This will exclude any possibility of a direct proton transfer between the dyad. The proposed mechanism of Miscione et al. [
83] is composed of three kinetic steps (relative to the formation of M1, M2, and M3).
4. BH3-Only Protein Noxa
4.1. Discovery
In 1990, Noxa was first identified as a cDNA clone during a screen in adult T-cell leukemia cells for gene products involved in tumorigenesis by Hijikata et al. [
93]. When peripheral blood mononuclear, human embryonic lung, and Jurkat T acute lymphoblastic leukemia cells were treated with the tumor promoter mitogen phorbol-12-myristate-13-acetate (PMA) Hijikata et al. [
93] observed a rapid induction of a novel transcript, which he named ATL-derived PMA-responsive gene. Under the HUGO system, it was later termed PMA-induced protein 1 (PMAIP1). [
94].
4.2. General Features and Transcript Variants
Human Noxa encodes a 54-amino acid protein that contains a single Bcl-2 homology 3 (BH3) domain [
95] and a C-terminal MTD (stands for mitochondrial targeting domain) [
96] that are both conserved between multiple mammalian species. The core gene structure of human Noxa contains three exons and two introns [
97]. To date, three splice variants of human Noxa have been identified. Transcript 1, which includes exon 1 and 3, encodes for the 54 amino acid Noxa where both the BH3 domain and MTD are encoded within exon 3.
Transcripts 2 and 3, named Noxa-splicing variants 1 and 2, respectively (
NSV-1/2), both contain exons 1 and 3, although
NSV-1 contains a portion of exon 2 (2a), and
NSV-2 contains the entire exon 2 [
97]. Sequence analysis of
NSV-1 and
NSV-2 predict the synthesis of 136 amino acid and 70 amino acid proteins respectively [
97]. Both
NSV-1/2 lack BH3 domains, due to differences in the reading frame of
NSV-1/2 as compared to Noxa, and have extremely short protein half-lives, as they are undetectable without treatment with the proteasome inhibitor MG132 [
97]. The in vivo function of these Noxa splice variants is still to be determined.
4.3. Regulation of Noxa Expression and Post-Translational Modification
Early observations indicated that Noxa transcription was primarily induced by p53. In wild-type and IRF-1-deficient MEFs, X-ray irradiation caused rapid induction of Noxa mRNA, whereas no Noxa induction was observed in p53¯/¯ MEFs [
95]. Analysis of the Noxa promoter region revealed a bona fide p53-response element 195 bp upstream of the transcription start site [
95,
96]. Additional studies have investigated p53-dependent transcription of Noxa either by in situ hybridization in p53¯/¯ mice or treatment of multiple cell lines with various chemical compounds [
98,
99,
100,
101]. The p53 independent regulation of Noxa by various stimuli has also been observed. Noxa induction was observed when multiple p53¯/¯ melanoma cell lines, along with PC-3 prostate cells and Saos-2 osteosarcoma cells (both p53 null cell lines) were treated with the γ-secretase inhibitor GSI [
102]. Hypoxia-induced HIF-1α has been shown to induce Noxa mRNA and protein expression in H719 and Saos-2 cells independent of p53 by binding to a hypoxia response element (HRE), at −1275 bp, within the Noxa promoter [
60]. Overexpression of adenovirus E1A protein, in the neuroblastoma cell line SH-SY5Y (non-functional p53) and SaOS-2 cells, results in activation of p73 and induction of Noxa mRNA [
103]. H
2O
2-induces activating transcription factor 4 can induce Noxa mRNA expression in Jurkat cells by binding to a cAMP response element-binding site within the Noxa promoter [
104]. A FoxO-binding site has been identified within the Noxa promoter by treatment of Jurkat cells with α-tocopheryl succinate, resulting in activation of FoxO1 and FoxO1-mediated transcription of Noxa [
105]. Additional studies have been completed that have investigated p53-independent regulation of Noxa transcription [
106,
107,
108,
109,
110,
111,
112].
In addition to transcriptional regulation of Noxa, proteasomal degradation has been implicated in the control of Noxa protein stability. Noxa has a short half-life [
97], although it does not contain any PEST or known E3-ligase binding domains. KLF6-SV1 was observed to bind to Noxa and lead to its HDM2-mediated proteasomal degradation upon KLF6-SV1 overexpression in SKOV3 cells [
113]. Additionally, proteasome inhibition in SKOV3 cells by MG132 causes an increase in both KLF6-SV1 and Noxa [
113]. Treatment of MM.1S cells with the novel proteasome inhibitor MLN2238 results in increased expression of both p53 and Noxa [
114]. Treatment with the proteasome inhibitor bortezomib (PS-441, Velcade) results in Noxa mRNA and protein induction in both p53 wild-type and p53-null melanoma cells, but not in normal melanocytes [
115]. This effect of bortezomib was also observed in vivo and multiple other cell lines [
116]. A cellular myelocytomatosis viral oncogene (c-MYC) binding site within the Noxa promoter has been identified, and siRNA knockdown of c-MYC reduced bortezomib-induced Noxa mRNA expression in multiple melanoma cell lines, MDA-MB-231 cells, and HeLa cells [
117,
118]. Treatment of LX-2 cells (human hepatic stellate cells) with MG132 resulted in increased expression of both Noxa mRNA and Noxa protein, and the MG132-induced apoptosis in LX-2 cells was shown to require Noxa [
119].
4.4. Subcellular Localization and Association with Bcl-2-like Proteins
Assessment by immunostaining has shown that overexpressed mouse Noxa preferentially localizes to the mitochondria and that mutations within either the BH3 domain or MTD prevent mitochondrial localization [
95,
96]. Furthermore, Noxa constructs that are missing either the BH3 domain or MTD fail to induce apoptosis [
96]. This suggests that both the BH3 domain and MTD are required for Noxa-induced apoptosis due to the proximity of the BH3 domain and MTD mutations in either domain may change the overall conformation of Noxa and impair Mcl-1 binding [
94].
Noxa contains only a single BH3 domain, placing it in the growing category of pro-apoptotic BH3-only proteins. Studies have shown that overexpression of Noxa can significantly induces apoptosis in various cell lines [
95,
96,
116], as well as correlates with MOMP, reactive oxygen species (ROS) accumulation, and cytochrome
c release [
92,
96,
125].
The specificity of Noxa towards Mcl-1 and A1 is dependent on key amino acid residues within the Noxa BH3 domain. Mutations in the Noxa BH3 domain (m3) allow Noxa to bind to Bcl-xL with a 100-fold increased affinity versus wild-type Noxa and is a more potent inducer of apoptosis [
67], while other mutations within the BH3 domain rendered Noxa inactive [
95,
96]. Additionally, Bims chimeras containing the Noxa BH3 domain showed reduced apoptotic ability, as compared to the wild-type Bims, which have a high apoptotic potential. Bims-Noxa BH3 chimeras were also restricted to binding Mcl-1, further demonstrating how the Noxa BH3 domain controls binding specificity and apoptotic potential of Noxa [
67]. Interaction of Noxa with Mcl-1 has also been observed in melanoma cells treated with bortezomib [
135], and in MDN and Jurkat cells where endogenous Noxa/Mcl-1 complexes were detected [
106,
136].
Due to the specificity of Noxa for both Mcl-1 and A1 [
67], the cellular levels of Mcl-1 and A1 control sensitivity to Noxa-induced apoptosis. Overexpression of Noxa in MEFs leads to Mcl-1 degradation without significant induction of apoptosis [
66]. Consistent with the idea that Noxa needs to be complemented by Bad, which targets Bcl-xL, Bcl-2, and Bcl-w, to induce apoptosis [
67], overexpression of both Noxa and Bad was shown to induce apoptosis in MEFs [
66,
133]. Overexpression of Noxa in Bcl-xL¯/¯ MEFs induced Bak-dependent apoptosis, demonstrating that Mcl-1 and Bcl-xL constrain Bak and that Noxa specifically engages Mcl-1 to promote Bak-dependent apoptosis [
66]. Overexpression of Noxa has also been shown to disrupt Mcl-1/Bak complexes in multiple myeloma and B-cell lymphomas and in Jurkat cells [
140]. Noxa has also been shown to disrupt Mcl-1/Bim complexes in bortezomib-treated MDN cells [
136].
5. BH3-Only Protein Puma
Puma, a p53 Up-regulated Modulator of apoptosis protein, was first discovered and cloned as a transcriptional target of p53 by two independent laboratories 19 years ago [
142]. In the same year, Han and colleagues identified the bbc3 (Bcl-2 binding component 3) gene that corresponds to the Puma cDNA [
143]. Puma is a highly efficient pro-apoptotic protein, thought to be one of the most powerful and effective “killers” among the BH3-only proteins. The bbc3 gene has been reported to encode 4 different forms (α, β, γ, and δ) of which only the α and β forms contain the BH3 domain and thus display the pro-apoptotic activity. The length of the α Puma transcript is 1.6–1.9 kb encoding a 193 amino acid protein [
142]. This protein is highly conserved among vertebrate species, yet shows no significant homologies to any other known proteins aside from those with the BH3 domain [
52].
5.1. Regulation of the BH3-Only Protein Puma
Regulation of the Bcl-2 family occurs through distinct cytotoxic stimuli in a variety of ways, including enhanced transcription and post-translational modifications [
146]. Importantly, Puma mRNA is induced by p53-dependent and p53-independent apoptotic stimuli in several cancer cell lines [
147]. These results support the idea that the regulation of Puma mRNA levels and thus the pro-apoptotic activity of the protein represents a common target in different cell death pathways [
52,
143]. The complexity of Puma function results from this protein’s involvement with a vast number of physiological and pathological processes, including the immune response, cancer, and neurodegenerative diseases as well as bacterial and viral infections [
11,
145]. Regulation of Puma expression during programmed cell death is coordinated by different transcription factors, most notably p53 but also through the activity of several other transcription factors including p73, sp1, Fox03a, E2f1, CHOP, TRB3, AP-1, and c-Myc [
52,
145].
5.2. p53-Dependent Apoptosis
The Puma gene is a direct transcriptional target of the tumor suppressor p53 [
54]. The mutual interaction between p53 and Puma is an efficient mechanism for preventing the growth and division of abnormal cells, thereby protecting against the development of cancer [
148]. It is known that p53 is required for the induction of Puma in response to DNA damage, but can also act on Puma in response to oxidative stress, deficiency of growth factors, or viral infection [
52]. More so, a lack of Puma expression is often associated with the mutation or deletion of p53 function, which contributes to over 50% of human cancers [
71,
149]. Furthermore, p53 acts as a sensor of cell stress, responsible for tumor growth inhibition by either cell cycle arrest followed by DNA repair or by causing apoptosis through activating the transcription of several pro-apoptotic genes, including Puma. p53-dependent regulation of pro-apoptotic Puma expression and subsequent apoptosis relies on the functioning of GSK-3 (Glycogen synthase kinase-3) and acetyltransferase Tip60, which control the choice between cell cycle arrest and apoptosis [
150].
5.3. p53-Independent Apoptosis
Stimuli from stressed or damaged cells can up-regulate Puma expression either by p53-mediated activation or by other transcription factors [
6]. Puma plays a very important role in p53-independent apoptosis involved in the removal of damaged cells during hypoxia, infection, and cytokine or growth factor depletion. These conditions are strong signals for apoptosis, which can lead to irreversible damage in cells and tissues [
52,
151]. During such pathological conditions, induction of Puma mRNA expression and activity level is due to the activity of other transcription factors, such as p73, Sp1, or Fox03a depending on the cell types [
6,
52,
152]. Although the mechanism remains unknown, the regulation of Puma occurs mainly without the participation of p53 in compromised cells [
153].
Both p53-dependent and p53-independent inductions of apoptosis via Puma are involved in the immune response after bacterial and viral infections [
71,
153,
154]. The immune response starts with increased T cell proliferation but once the pathogen has been eliminated, the number of T cells needs to be controlled through apoptosis to decrease the immune response. Puma plays a role in T cell apoptosis and is driven both by p53 and Fox03a [
52,
155]. This ensures the proper functioning of the immune system to prevent pathological conditions, such as autoimmunity [
155].
This entry is adapted from the peer-reviewed paper 10.3390/life12020256