


Thiazole, a five-membered heteroaromatic ring, is an important scaffold of a large number of synthetic compounds. Its diverse pharmacological activity is reflected in many clinically approved thiazole-containing molecules, with an extensive range of biological activities, such as antibacterial, antifungal, antiviral, antiparasitic, antitumor, antiparkinsonian, and anti-inflammatory effects.
- thiazole
- bisthiazole
- synthesis
- derivatives
1. Introduction
Nitrogen-containing heterocyclic compounds play an important role in the drug discovery process, as approximately 75% of FDA (Food and Drug Administration)-approved small-molecule drugs contain one or more nitrogen-based heterocycles [1]. Thiazole, or 1,3-thiazole, belongs to the class of azoles and contains one sulfur atom and one nitrogen atom at positions 1 and 3. Its diverse biological activity is reflected in a large number of clinically approved thiazole-containing compounds with an extensive range of pharmacological activities. Most of these compounds are 2,4-disubstituted thiazole derivatives, and only a few are 2,5-disubstituted or 2,4,5-trisubstituted thiazoles [2].
Several drugs such as sulfathiazole; aztreonam; numerous cephems (ceftaroline, cefotiam, ceftibuten, cefixime, ceftriaxone, cefotaxime, ceftazidime, cefmenoxime, ceftizoxime, cefepime, cefdinir) with antibacterial effects; pramipexole with antiparkinsonian activity; edoxaban with antithrombotic effects; isavuconazole with antifungal effects; famotidine and nizatidine with antiulcer activity; meloxicam with anti-inflammatory effects; tiazofurin, dabrafenib, dasatinib, ixabepilone, and epothilone with antitumor effects; mirabegron as a β3-adrenergic receptor agonist; nitazoxanide and thiabendazole with antiparasitic effects; and febuxostat with antigout activity contain one thiazole moiety in the structure (Figure 1) [2][3][4].
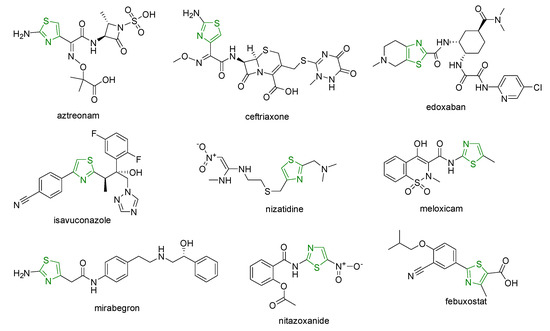
Figure 1. Clinical drugs bearing one thiazole ring.
Compounds bearing two thiazole rings, such as the antitumor drug bleomycin, the antiretroviral agent ritonavir, the pharmacokinetic enhancer for HIV drugs cobicistat, and the antibacterial agent cefditoren, have been authorized on the pharmaceutical market (Figure 2) [2][5].
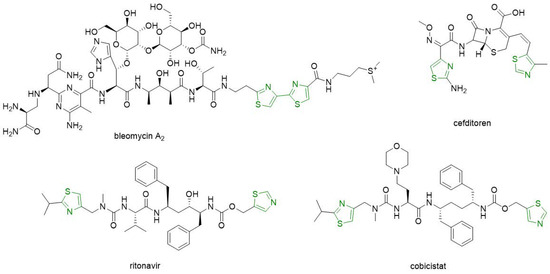
Figure 2. Clinical drugs bearing two thiazole rings.
The high medicinal significance of this scaffold has attracted considerable attention from many researchers and encouraged the design and synthesis of numerous thiazole- and bisthiazole-containing compounds with diverse pharmacological activities, such as antibacterial [6], antifungal [7], antiprotozoal [8], antiviral [9][10], anticancer [11], anti-inflammatory [12][13][14][15][16], antioxidant [17], analgesic [18], anticonvulsant [19], antidiabetic [20], and antihypertensive [21] activities. Furthermore, thiazole compounds exhibiting a promising biological potential for the treatment of Alzheimer’s disease [22][23] and metabolic syndrome [24] have been reported in the scientific literature.
Nowadays, research in the field of antimicrobial drug design is focused on the discovery of novel targets and chemical entities that possess antibacterial activity in order to overcome the rapid development of drug resistance. The few antibiotics that have been recently approved are structurally related to older drugs, being susceptible to the same mechanisms of resistance [25]. Tropical diseases such as Chagas disease, leishmaniasis, and malaria occur especially in underdeveloped countries, around 266 million cases being reported globally [26]. At the present time, only a small number of drugs are available on the pharmaceutical market for the treatment of protozoan infections [27]. Cancer remains one of the leading causes of mortality worldwide. The lack of selectivity and target specificity, as well as the presence of toxicity and resistance, is an inadequate feature of the currently available anticancer drug therapy [3]. Considering all the above, there is an urgent need for continuous progress in the design and development of anti-infective and antitumor agents.
2. Chemistry of Thiazole
Free thiazole is a pale-yellow flammable liquid, with a pyridine-like odor and a boiling point in the range of 116–118 °C. It has an aromatic character, due to the delocalization of a lone pair of electrons from the sulfur atom, resulting in a 6π-electron system. Also, its high aromaticity is highlighted by proton nuclear magnetic resonance, the chemical shift values of each proton within the thiazole ring being situated between 7.27 and 8.77 ppm. The resonance structures of thiazole are illustrated in Figure 3 [28][29].
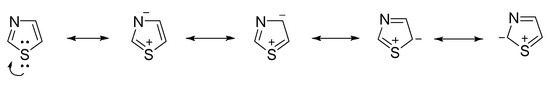
Figure 3. The resonance structures of thiazole.
The calculated π-electron density revealed that the electrophilic substitution takes place preferentially at the C-5 position, followed by the C4-position (Figure 4). The nucleophilic substitution occurs at the C-2 position [30].
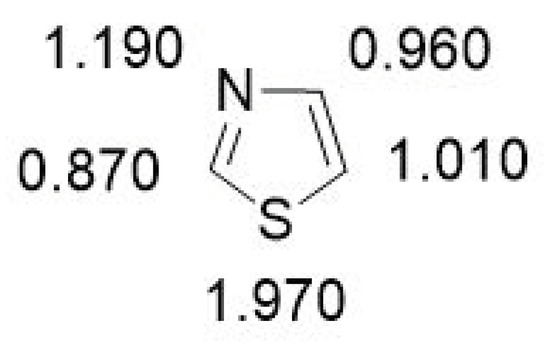
Figure 4. Calculated π-electron density of thiazole.
The acidity given by the presence of the three hydrogen atoms decreases in the order H2 >> H5 > H4 [4].
3. Synthesis of Thiazole and Bisthiazole Derivatives
Hantzsch synthesis is the oldest and most widely known method for the synthesis of a thiazole ring. The method consists of a cyclization reaction between alpha-halocarbonyl compounds and various reactants containing the N-C-S fragment. Examples of such compounds include thiourea, thioamides, thiosemicarbazides, and thiosemicarbazones [31].
The condensation of thioamides with various alpha-halocarbonyl compounds is commonly used. Many thiazoles with alkyl, aryl, or heteroaryl substituents at position 2, 4, or 5 can be obtained through this reaction. The reaction mechanism consists of the nucleophilic attack of the thioamide sulfur atom on the alpha carbon of the alpha-halocarbonyl, with the formation of an intermediate compound, which by subsequent dehydration leads to the corresponding thiazole (Scheme 1).
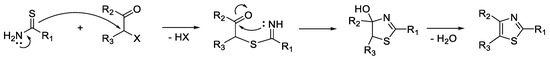
Scheme 1. Reaction mechanism of Hantzsch thiazole synthesis.
By the reaction of thiourea with alpha-halocarbonyl compounds, monosubstituted or disubstituted 2-aminothiazoles can be obtained, while by using other compounds containing thioamide moieties, such as thiosemicarbazides and thiosemicarbazones, 2-hydrazinothiazole and thiazol-2-yl-hydrazone derivatives can be synthesized in good yields. The condensation reactions occur through imino thioether and hydroxythiazoline intermediates, which are sometimes stable and isolable. The alpha-halocarbonyl component may be represented by alpha-haloketones and alpha-haloesters [32][33].
Another method of obtaining thiazoles is represented by Gabriel synthesis, which consists of the cyclization reaction of acylaminocarbonyl compounds and a stoichiometric amount of phosphorus pentasulfide at 170 °C (Scheme 2) [28][34].
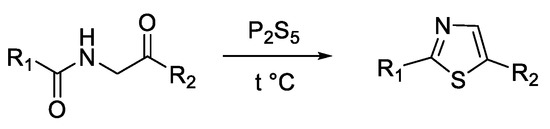
Scheme 2. Gabriel thiazole synthesis.
Cook–Heilbron synthesis leads to 2,4-disubstituted 5-aminothiazole derivatives by the reaction of an α-aminonitrile and dithioacids or esters of dithioacids, carbon disulfide, carbon oxysulfide, or isothiocyanates under mild reaction conditions (Scheme 3). When carbon disulfide is used in the reaction, 5-amino-2-mercaptothiazole compounds are formed (Scheme 4) [32][33].
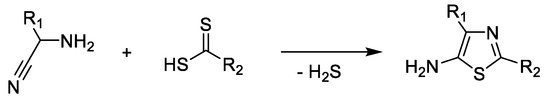
Scheme 3. Cook–Heilbron synthesis of 5-aminothiazoles.
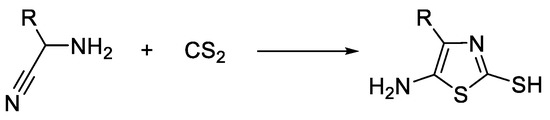
Scheme 4. Cook–Heilbron synthesis of 5-amino-2-mercaptothiazoles.
Lingaraju et al. [35] efficiently synthesized a series of 4,5-disubstituted thiazole derivatives from active methylene isocyanides and methyl carbodithioates (Scheme 5). The reaction took place in the presence of a strong base, sodium hydride, and dimethylformamide (DMF) as a solvent. The short reaction time (10–30 min) and obtaining sufficiently pure final products in good yields are the main advantages of this method. Moreover, this procedure made it possible to obtain 2-unsubstituted thiazoles, which cannot be easily synthesized through other approaches such as Hantzsch or Cook–Heilbron synthesis.
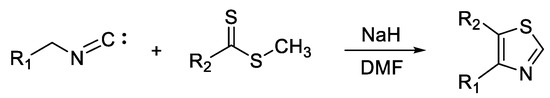
Scheme 5. Synthesis of thiazoles from isocyanide derivatives and carboxymethyl dithioates.
The proposed mechanism is illustrated in Scheme 6. Carbanion 2, formed by removing a proton from the active methylene group of compound 1, in the presence of sodium hydride, attacks dithioester 3, generating an unstable intermediate 4. Subsequently, in the presence of sodium hydride, carbanion 5 is obtained, being in equilibrium with the enethiolate anion 6. In the end, thiazole nucleus 7 is formed following the intramolecular cyclization of compound 6.
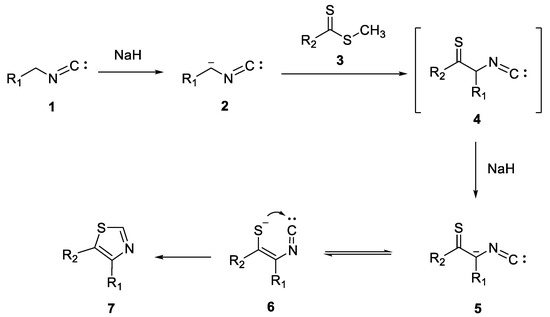
Scheme 6. The proposed mechanism for the synthesis of 4,5-disubtituted thiazole derivatives.
Castagnolo et al. [36] reported the synthesis of 2-aminothiazoles by a domino alkylation-cyclization reaction starting from various substituted propargyl bromides and thiourea derivatives. The synthesis was carried out in the presence of a stoichiometric amount of potassium carbonate and DMF as a solvent, at a temperature of 130 °C, under microwave irradiation, for 10 min (two cycles of 5 min each), as illustrated in Scheme 7.
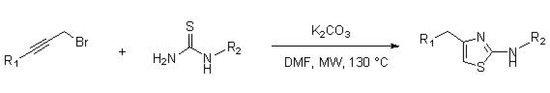
Scheme 7. Synthesis of 2-aminothiazoles from substituted propargyl bromide and thioureas.
This method is attractive due to the high availability of alkynes, which are an important alternative to α-haloketones used in Hantzsch synthesis, the main disadvantage of the latter being the strongly irritating character.
The proposed reaction mechanism is shown in Scheme 8. The initial step consists of the alkylation of thiourea or substituted thioureas, leading to the formation of intermediate 8. Subsequently, compound 8 undergoes a 5-exo-dig cyclization reaction with the generation of intermediate 9, which is transformed through an isomerization reaction into the final reaction product, 2-aminothiazole 10.
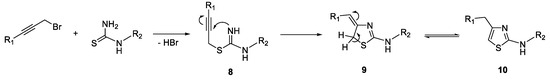
Scheme 8. The proposed mechanism for the synthesis of 2-aminothiazole derivatives.
Tang et al. [37] developed a method of obtaining thiazoles by a copper-catalyzed cyclization reaction starting from oximes, anhydrides, and potassium thiocyanate (Scheme 9). To optimize reaction conditions, the authors examined various solvents, such as 1,2-dichloroethane, 1,4-dioxane, acetonitrile and toluene, different copper salts (such as CuI, CuCl, CuBr, CuBr2, Cu(OAc)2, and Cu(OTf)2 and their replacement with other metal salts such as Fe(OAc)2, FeBr2, FeBr3, FeCl3, PdCl2, and AgCl), and different sulfur sources such as KSCN, NaSCN, NH4SCN, S, and Na2S.
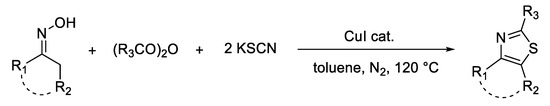
Scheme 9. Synthesis of thiazoles from oximes, anhydrides, and KSCN.
Good to excellent yields (up to 85%) were obtained when toluene was used as a solvent, copper(I) iodide as a catalyst, and two equivalents of KSCN as a sulfur source. The reaction took place at a temperature of 120 °C, under a nitrogen atmosphere, for 24 h.
Wang et al. [38] obtained a series of thiazole derivatives starting from aldehydes, amines, and element sulfur through an oxidation reaction catalyzed by copper salts in the presence of molecular oxygen (Scheme 10). The main advantages of this method are the high availability of the starting materials, the low-cost catalyst, and the use of a green oxidant.
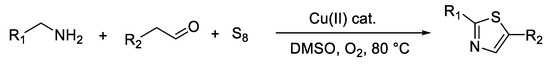
Scheme 10. Synthesis of thiazoles from aldehydes, amines, and element sulfur.
Chen et al. [39] successfully synthesized 4-substituted 2-aminothiazoles and 4-substituted 5-thiocyano-2-aminothiazoles starting from substituted vinyl azides and potassium thiocyanate under different reaction conditions. They found that the use of palladium(II) acetate as a catalyst and n-propanol as a solvent led to 4-substituted 2-aminothiazoles, after a reaction time of 12 h, at 80 °C, while in the presence of iron(III) bromide as a catalyst and acetonitrile as a solvent, 4-substituted 5-thiocyano-2-aminothiazoles were formed (Scheme 11).
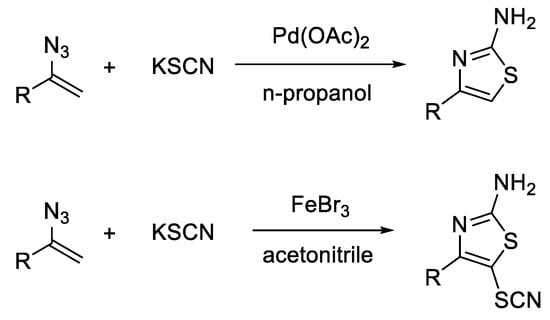
Scheme 11. Synthesis of thiazoles from vinyl azides and potassium thiocyanate under different reaction conditions.
When palladium(II) acetate was used as a catalyst, vinyl azides could be substituted with either aromatic (phenyl, naphthyl) or alkyl substituents, while the reaction catalyzed by iron(III) bromide took place only when vinyl azides were substituted with aromatic residues. In both cases, if the phenyl residue is substituted with an electron-withdrawing group (-NO2, -COOCH3), the reaction yield decreases. Moreover, if an electron-withdrawing group, such as an ester or a benzoyl group, binds directly to the alpha-carbon of vinyl azide, the reaction no longer takes place.
The study of the mechanism showed that the reaction occurred through an ionic mechanism, in the presence of palladium(II) acetate, and through a radical mechanism, in the presence of iron(III) bromide, thus explaining the formation of different reaction products [39].
Numerous heterocyclic compounds possessing the thiazole ring have been obtained in good to excellent yields using microwave irradiation [40][41]. Compared to conventional methods, microwave-assisted synthesis has several advantages, being an environmentally friendly and cost-effective tool, that leads to improved yields in short reaction times [42].
Asif et al. [43] developed a simple, one-pot, efficient method for the synthesis of novel steroidal thiazole derivatives through the condensation reaction of 2-bromoacetophenone, thiosemicarbazide, and steroidal carbonyl compounds (Scheme 12). The reaction took place under microwave heating at 60 °C, in ethanol, for 35–45 min, obtaining the target compounds in good yields (80–85%).
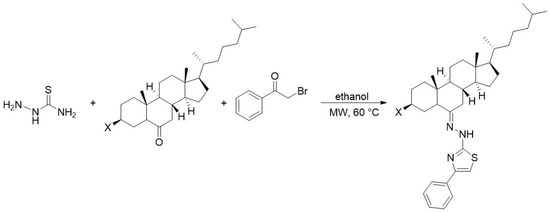
Scheme 12. Synthesis of thiazoles from thiosemicarbazide, ketones, and 2-bromoacetophenone under microwave irradiation.
Mamidala et al. [44] reported the synthesis of new coumarin-based thiazole derivatives, starting from thiocarbohydrazide, aldehydes, and α-halocarbonyl coumarins in a molar ratio of 1:2:1, under microwave heating (Scheme 13). Various solvents like methanol, ethanol, dimethyl sulfoxide (DMSO), and acetonitrile and different catalysts such as acetic acid, sulfuric acid, and hydrochloric acid were investigated in order to optimize the reaction regarding time and yield. Thus, it was observed that using ethanol as a solvent and a catalytic amount of acetic acid under microwave irradiation (70 °C, 210 W) led to high yields (88–93%) and short reaction times (5–8 min).
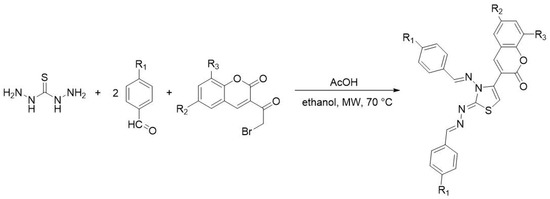
Scheme 13. Synthesis of thiazoles from thiocarbohydrazide, aldehydes, and α-bromoketones under microwave irradiation.
Chinnaraja and Rajalakshmi [45] reported the microwave-assisted synthesis of novel hydrazinyl thiazole derivatives within 30–175 s under solvent- and catalyst-free conditions. Starting from either various carbonyl compounds, thiosemicarbazide, and alpha-haloketones or substituted thiosemicarbazones and alpha-haloketones (Scheme 14), the target compounds were obtained in good to excellent yield and high purity through an eco-friendly, one-pot procedure.
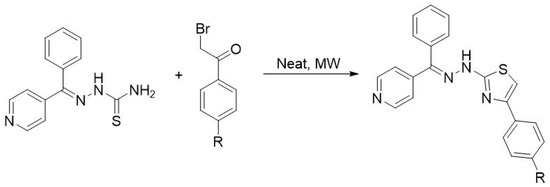
Scheme 14. Synthesis of thiazoles from thiosemicarbazones and α-bromoketones under microwave irradiation.
References
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909.
- Scott, K.A.; Njardarson, J.T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top. Curr. Chem. 2018, 376, 5.
- Sharma, P.C.; Bansal, K.K.; Sharma, A.; Sharma, D.; Deep, A. Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur. J. Med. Chem. 2020, 188, 112016.
- Pola, S. Significance of Thiazole-based Heterocycles for Bioactive Systems. In Scope of Selective Heterocycles from Organic and Pharmaceutical Perspective; Varala, R., Ed.; InTech: Rijeka, Croatia, 2016; pp. 1–47.
- Gallant, J.E.; Koenig, E.; Andrade-Villanueva, J.; Chetchotisakd, P.; DeJesus, E.; Antunes, F.; Arastéh, K.; Moyle, G.; Rizzardini, G.; Fehr, J.; et al. Cobicistat Versus Ritonavir as a Pharmacoenhancer of Atazanavir Plus Emtricitabine/Tenofovir Disoproxil Fumarate in Treatment-Naive HIV Type 1–Infected Patients: Week 48 Results. J. Infect. Dis. 2013, 208, 32–39.
- Mishra, I.; Mishra, R.; Mujwar, S.; Chandra, P.; Sachan, N. A retrospect on antimicrobial potential of thiazole scaffold. J. Heterocycl. Chem. 2020, 57, 2304–2329.
- Meleddu, R.; Distinto, S.; Corona, A.; Maccioni, E.; Arridu, A.; Melis, C.; Bianco, G.; Matyus, P.; Cottiglia, F.; Sanna, A.; et al. Exploring the thiazole scaffold for the identification of new agents for the treatment of fluconazole resistant Candida. J. Enz. Inhib. Med. Chem. 2016, 31, 1672–1677.
- da Silva, E.B.; Oliveira e Silva, D.A.; Oliveira, A.R.; da Silva Mendes, C.H.; dos Santos, T.A.R.; da Silva, A.C.; de Castro, M.C.A.; Ferreira, R.S.; Moreira, D.R.M.; de Oliveira Cardoso, M.V.; et al. Desing and synthesis of potent anti-Trypanosoma cruzi agents new thiazoles derivatives which induce apoptotic parasite death. Eur. J. Med. Chem. 2017, 130, 39–50.
- Singh, I.P.; Gupta, S.; Kumar, S. Thiazole Compounds as Antiviral Agents: An Update. Med. Chem. (Los. Angeles) 2020, 16, 4–23.
- Abu-Melha, S.; Edrees, M.M.; Riyadh, S.M.; Abdelaziz, M.R.; Elfiky, A.A.; Gomha, S.M. Clean grinding technique: A facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing thiazole moiety against SARS-CoV-2 main protease (Mpro). Molecules 2020, 25, 4565.
- Gomha, S.; Edrees, M.; Altalbawy, F. Synthesis and Characterization of Some New Bis-Pyrazolyl-Thiazoles Incorporating the Thiophene Moiety as Potent Anti-Tumor Agents. Int. J. Mol. Sci. 2016, 17, 1499.
- Woods, K.W.; McCroskey, R.W.; Michaelides, M.R.; Wada, C.K.; Hulkower, K.I.; Bell, R.L. Thiazole analogues of the NSAID indomethacin as selective COX-2 Inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1325–1328.
- Araniciu, C.; Pârvu, A.; Palage, M.; Oniga, S.; Benedec, D.; Oniga, I.; Oniga, O. The Effect of Some 4,2 and 5,2 Bisthiazole Derivatives on Nitro-Oxidative Stress and Phagocytosis in Acute Experimental Inflammation. Molecules 2014, 19, 9240–9256.
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685.
- Abdelall, E.K.A.; Kamel, G.M. Synthesis of new thiazolo-celecoxib analogues as dual cyclooxygenase-2/15-lipoxygenase inhibitors: Determination of regio-specific different pyrazole cyclization by 2D NMR. Eur. J. Med. Chem. 2016, 118, 250–258.
- Rödl, C.B.; Vogt, D.; Kretschmer, S.B.M.; Ihlefeld, K.; Barzen, S.; Brüggerhoff, A.; Achenbach, J.; Proschak, E.; Steinhilber, D.; Stark, H.; et al. Multi-dimensional target profiling of N,4-diaryl-1,3-thiazole-2-amines as potent inhibitors of eicosanoid metabolism. Eur. J. Med. Chem. 2014, 84, 302–311.
- Jaishree, V.; Ramdas, N.; Sachin, J.; Ramesh, B. In vitro antioxidant properties of new thiazole derivatives. J. Saudi Chem. Soc. 2012, 16, 371–376.
- Saravanan, G.; Alagarsamy, V.; Prakash, C.R.; Kumar, P.D.; Selvam, T.P. Synthesis of Novel Thiazole Derivatives as Analgesic Agents. Asian J. Pharm. Sci. 2011, 1, 134–138.
- Siddiqui, N.; Ahsan, W. Triazole incorporated thiazoles as a new class of anticonvulsants: Design, synthesis and in vivo screening. Eur. J. Med. Chem. 2010, 45, 1536–1543.
- Wang, G.; Peng, Z.; Gong, Z.; Li, Y. Synthesis, biological evaluation, and docking studies of novel 5,6-diaryl-1,2,4-triazine thiazole derivatives as a new class of α-glucosidase inhibitors. Bioorg. Chem. 2018, 78, 195–200.
- Sowjanya, C.; Seetaram Swamy, S.; Gomathi, S.; Ashok Babu, K. Synthesis, Chemistry and Anti-Hypertensive Activity of Some New Thiazole-Thiadiazole Derivatives. Int. J. Adv. Res. Med. Pharm. Sci. 2016, 1, 6–10.
- Sahin, Z.; Ertas, M.; Bender, C.; Bülbül, E.F.; Berk, B.; Biltekin, S.N.; Yurttaş, L.; Demirayak, Ş. Thiazole-substituted benzoylpiperazine derivatives as acetylcholinesterase inhibitors. Drug Dev. Res. 2018, 79, 406–425.
- Sagar, S.R.; Singh, D.P.; Das, R.D.; Panchal, N.B.; Sudarsanam, V.; Nivsarkar, M.; Vasu, K.K. Pharmacological investigation of quinoxaline-bisthiazoles as multitarget-directed ligands for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 89, 1–17.
- Kadam, K.S.; Jadhav, R.D.; Kandre, S.; Guha, T.; Reddy, M.M.K.; Brahma, M.K.; Deshmukh, N.J.; Dixit, A.; Doshi, L.; Srinivasan, S.; et al. Evaluation of thiazole containing biaryl analogs as diacylglycerol acyltransferase 1 (DGAT1) inhibitors. Eur. J. Med. Chem. 2013, 65, 337–347.
- Hoffman, P.S. Antibacterial Discovery: 21st Century Challenges. Antibiotics 2020, 9, 213.
- Scarim, C.B.; Jornada, D.H.; Machado, M.G.M.; Ferreira, C.M.R.; dos Santos, J.L.; Chung, M.C. Thiazole, thio and semicarbazone derivatives against tropical infective diseases: Chagas disease, human African trypanosomiasis (HAT), leishmaniasis, and malaria. Eur. J. Med. Chem. 2019, 162, 378–395.
- Müller, J.; Hemphill, A. Drug target identification in protozoan parasites. Expert Opin. Drug Discov. 2016, 11, 815–824.
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718.
- Sarangi, P.K.N.; Sahoo, J.; Swain, B.D.; Paidesetty, S.K.; Mohanta, G.P. Thiazoles as potent anticancer agents: A review. Indian Drugs 2016, 53, 5–11.
- Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles. Structure, Reactions, Syntheses, and Applications; WILEY-VCH GmbH & Co. KGaA: Weinheim, Germany, 2003; ISBN 3-527-30720-6.
- Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M. Novel thiazole derivatives: A patent review (2008-2012; Part 1). Expert Opin. Ther. Pat. 2014, 24, 201–216.
- Mishra, C.B.; Kumari, S.; Tiwari, M. Thiazole: A promising heterocycle for the development of potent CNS active agents. Eur. J. Med. Chem. 2015, 92, 1–34.
- Ali, S.H.; Sayed, A.R. Review of the synthesis and biological activity of thiazoles. Synth. Commun. 2020, 50, 1–31.
- Nayak, S.; Gaonkar, S.L. A Review on Recent Synthetic Strategies and Pharmacological Importance of 1,3-Thiazole Derivatives. Mini-Reviews Med. Chem. 2019, 19, 215–238.
- Lingaraju, G.; Swaroop, T.; Vinayaka, A.; Sharath Kumar, K.; Sadashiva, M.; Rangappa, K. An Easy Access to 4,5-Disubstituted Thiazoles via Base-Induced Click Reaction of Active Methylene Isocyanides with Methyl Dithiocarboxylates. Synthesis (Stuttg) 2012, 44, 1373–1379.
- Castagnolo, D.; Pagano, M.; Bernardini, M.; Botta, M. Domino Alkylation-Cyclization Reaction of Propargyl Bromides with Thioureas/Thiopyrimidinones: A New Facile Synthesis of 2-Aminothiazoles and 5H-Thiazolo[3,2-a]pyrimidin-5-ones. Synlett 2009, 2009, 2093–2096.
- Tang, X.; Yang, J.; Zhu, Z.; Zheng, M.; Wu, W.; Jiang, H. Access to Thiazole via Copper-Catalyzed [3+1+1]-Type Condensation Reaction under Redox-Neutral Conditions. J. Org. Chem. 2016, 81, 11461–11466.
- Wang, X.; Qiu, X.; Wei, J.; Liu, J.; Song, S.; Wang, W.; Jiao, N. Cu-Catalyzed Aerobic Oxidative Sulfuration/Annulation Approach to Thiazoles via Multiple Csp 3 –H Bond Cleavage. Org. Lett. 2018, 20, 2632–2636.
- Chen, B.; Guo, S.; Guo, X.; Zhang, G.; Yu, Y. Selective Access to 4-Substituted 2-Aminothiazoles and 4-Substituted 5-Thiocyano-2-aminothiazoles from Vinyl Azides and Potassium Thiocyanate Switched by Palladium and Iron Catalysts. Org. Lett. 2015, 17, 4698–4701.
- Gomha, S.M.; Edrees, M.M.; Faty, R.A.M.; Muhammad, Z.A.; Mabkhot, Y.N. Microwave-assisted one pot three-component synthesis of some novel pyrazole scaffolds as potent anticancer agents. Chem. Cent. J. 2017, 11, 37.
- Karamthulla, S.; Pal, S.; Khan, M.N.; Choudhury, L.H. “On-water” synthesis of novel trisubstituted 1,3-thiazoles via microwave-assisted catalyst-free domino reactions. RSC Adv. 2014, 4, 37889–37899.
- Prajapati, N.P.; Patel, K.D.; Vekariya, R.H.; Patel, H.D.; Rajani, D.P. Thiazole fused thiosemicarbazones: Microwave-assisted synthesis, biological evaluation and molecular docking study. J. Mol. Struct. 2019, 1179, 401–410.
- Asif, M.; Ali, A.; Zafar, A.; Farhan, M.; Khanam, H.; Hadi, S.M. Shamsuzzaman Microwave-assisted one pot synthesis, characterization, biological evaluation and molecular docking studies of steroidal thiazoles. J. Photochem. Photobiol. B Biol. 2017, 166, 104–115.
- Mamidala, S.; Peddi, S.R.; Aravilli, R.K.; Jilloju, P.C.; Manga, V.; Vedula, R.R. Microwave irradiated one pot, three component synthesis of a new series of hybrid coumarin based thiazoles: Antibacterial evaluation and molecular docking studies. J. Mol. Struct. 2021, 1225, 129114.
- Chinnaraja, D.; Rajalakshmi, R. A facile, solvent and catalyst free, microwave assisted one pot synthesis of hydrazinyl thiazole derivatives. J. Saudi Chem. Soc. 2015, 19, 200–206.
