Skin cancer is the out-of-control growth of abnormal skin cells [
1,
2]. It is one of the most frequent types of cancer, being responsible for over 19 million new cases of cancer and almost 10 million deaths worldwide in 2020 [
3]. Concerning its geographic distribution, North America is estimated to represent the majority of the cases (45.4%), followed by Europe (28.4%), Central and South America (9.5%), Australia, and New Zealand (5.8%). In Europe, with 150,627 new cases of melanoma and over 356,180 non-melanoma skin cancers. In 2022, melanoma caused 26,360 deaths, while for non-melanoma skin cancers there were 12,679 deaths, in Europe [
4]. The continuous incidence of skin cancer is due to several factors, being the main reason for ultraviolet light exposure [
5,
6,
7], but also due to environmental and hereditary risk factors [
8,
9]. At the pathophysiological level, it is divided into two groups: melanoma and non-melanoma tumors. While melanoma derives from melanocytes, non-melanoma tumors derive from cells from the epiderm. The most common skin tumors are basal cell carcinomas, the most frequent of them arising from interfollicular epidermis exhibiting mutations on TP53 gene; squamous cell carcinoma with origin from squamous keratinocytes in the epidermis of the skin or mucous membranes; and melanoma with origin from melanocytes present in the skin, which incidence every year is notably increasing. Particularly in the United States (U.S.), the economic burden of this disease was USD 8.9 billion in recent years (2016–2018). About USD 6.5 billion were related to non-melanoma tumors, whereas approximately USD 2.5 billion were associated with melanoma tumors. The latter is expected to triple by 2030 as a result of the overall rising of healthcare costs [
10]. The type of treatment chosen depends on tumor type, location, and progression degree. There is a wide variety of treatment options, starting with surgical treatment, such as excision surgery, Mohs surgery and curettage, and cryotherapy, radiotherapy, chemotherapy, and photodynamic therapy [
11,
12,
13,
14,
15]. Although there are already several treatment options available, its high incidence makes skin cancer an important health problem and relevant to the study and development of new approaches for skin cancer management [
1]. New therapies can include cancer immunotherapy, cancer photodynamic and/or photothermal therapy, and cancer photoimmunotherapy. More recently, new approaches, such as the use of metallic, polymeric, and lipid-based nanotheranostics, emerged in this field to facilitate the treatment and diagnosis of this disease. These platforms may serve as contrast agents with therapeutic properties (e.g., reactive oxygen species production). Moreover, the versatility of their formulations allows the incorporation of other materials (e.g., gold) and/or drugs (e.g., parvifloron D), which act synergistically to impair the development of solid and superficial tumors, namely at early stages [
16,
17].
1.1. Immunotherapy
Cancer immunotherapy has been contributing to improved survival and quality of life for cancer patients. It consists in triggering the immune system to control and fight cancer, overcoming the mechanisms that cancer cells develop to escape immune surveillance and avoid detection and elimination [
18,
19]. Immunotherapy started more than 100 years ago, in New York, with Dr. Coley, who treated sarcoma patients with Coley’s toxin, a vaccine with a mixture of two bacterial toxins [
20]. Currently, there are two types of immunotherapies, active and passive immunotherapy. Active immunotherapy stimulates the patient’s immune system, whereas passive immunotherapy can be with the administration of, for example, cytokines, vaccines, and antibodies [
21,
22,
23].
The immune system plays a critical role in recognizing, eliminating, and controlling tumor progression. However, cancer cells develop mechanisms to avoid it, namely: (i) downmodulation of components of antigen processing and presentation machinery; (ii) an environment that promotes suppressor immune cells, such as regulatory T cells (Treg), an immunosuppressive subset of CD4+ T-cell family, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages, which are anti-inflammatory macrophages (M2- like); (iii) production of soluble factors associated with immunosuppression, such as TGF-β and IL-10; (iv) and upregulation of ligands for coinhibitory receptors that downmodulate programmed death ligand-1 (PD-L1) [
24,
25,
26,
27,
28,
29].
Dendritic cells (DCs) induce the differentiation of T cells to their antigen-specific effector T cells. CD4+ T cells are responsible for inducing DC maturation and for CD8+ T-cell priming. The primed cells are activated to form cytotoxic T lymphocytes, and these are responsible for releasing INF-γ and TNF-α, which will induce cytotoxicity in cancer cells. INF-γ is produced by both CD4+ and CD8+ T cells and stimulates the antitumor pro-inflammatory macrophages (M1). These tumor suppressor cells, such as cytotoxic T lymphocytes, also upregulate the release of pro-inflammatory cytokines, namely, IL-2, IL-6, IL-12, INF-γ, and TNF-α [
24,
30,
31,
32]. The higher calreticulin (CRT) exposure and release of high mobility group box 1 (HMGB-1) also act as an “eat-me” signal to induce tumor cell apoptosis [
33,
34,
35,
36].
1.2. Phototherapy for Cancer Treatment
Phototherapy (PT) started 4000 years ago in ancient Egypt to treat Vitiligo, when a plant extract was boiled and then combined with sun exposure. Modern phototherapy, on the other hand, started only in the 70s, using artificial light sources [
37,
38,
39]. Phototherapy is mainly divided into two categories, photodynamic therapy (PDT) and photothermal therapy (PTT). In PDT, a photosensitizer agent is irradiated by light to generate reactive oxygen species (ROS). These are highly toxic, causing cell death. PTT is based on local temperature increase, usually triggered by laser radiation. Usually, NIR lasers (650–1350 nm) are used in PTT due to their efficiency in penetrating tumors [
40,
41,
42,
43]. PTT can be divided into two categories. In the first, called mild hyperthermia, temperature increases up to 43–50 °C, leading to enhanced membrane permeability, cellular uptake, metabolic signaling disruption, and dysfunctional membrane transport. The capability of tumor cells to recover from such damages is very low. The second one is photothermal ablation (>50 °C), which destroys the cellular membrane, leading to necrotic cell death [
44]. PDT has been FDA-approved for almost 40 years [
40,
45]. Hematopotphyrin derivative (HPD) was the first PS receiving FDA approval, nowadays Foscan
®, Levulan
®, Radachlorin
®, Metvix
®, and Photofrin
® are FDA-approved PS [
39,
43].
1.3. Photoimmunotherapy for Cancer Treatement
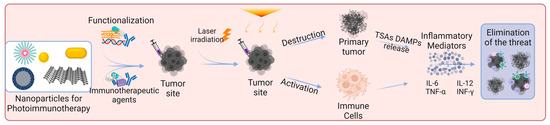
PT triggers immunogenic cell death (ICD) that will release tumor-specific antigens (TSAs) and damage-associated molecular patterns (DAMPs), namely CRT, HMGB-1, and ATP. This phenomenon increases the immunogenicity of the tumor microenvironment once DAMPs induce the maturation of DCs, and pro-inflammatory cytokines, such as IL-2, IL-6, IL-12, INF-γ and TNF-α, were also reported to increase. The immunostimulatory effect of PT boosts anti-tumor immunity when compared to immunotherapy alone. Although immunotherapy by itself can be effective in triggering the immune response at tumor site, it is inefficient to eradicate primary tumors [
46,
47,
48,
49,
50].
When combining phototherapy with immunotherapy, in photoimmunotherapy (PIT), a synergy is reported to occur between them. PT directly kills the tumor cells and triggers a systemic immune response, and when in combination with immunotherapy, immunological memory is formed. Photoimmunotherapy has the advantages of phototherapy and the ability to trigger an immune response, making it ideal for treating metastatic cancer. Thereby, PIT eradicates primary tumors and, through simultaneously stimulating immune memory, it has the potential to prevent tumor recurrence and metastasis [
42,
46,
51,
52].
2. Nanomaterials
2.1. Structure and Properties
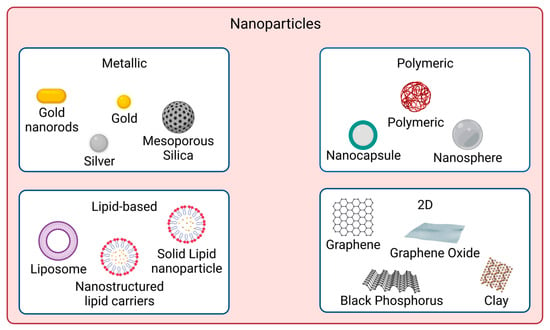
In recent years, nanomaterials have gained much interest in the biomedical field, namely in drug delivery, tissue engineering, diagnosis, and theragnostics, amongst others. Nanomaterials can be divided into different categories according to their properties (e.g., size, shape, physicochemical properties, etc.). Regarding nanomaterials used for skin cancer photoimmunotherapy, the focus of this review, the main categories found are metallic, polymeric, lipid-based and 2D nanomaterials [
53], as illustrated in
Figure 1.
Figure 1. Nanomaterial types used for skin cancer photoimmunotherapy. The main categories are metallic, polymeric, lipid-based, and 2D nanoparticles. Created with BioRender.com.
Metallic nanoparticles are nanosized metals which present a metal core covered in a metal shell. Their size can range from 1 up to 100 nm. Surface modification strategies are employed on metallic nanoparticles to prevent their agglomeration in physiological conditions. Nanoparticles with a zeta potential greater than 30 mV and lower than −30 mV are more water stable. Additionally, particle shape is crucial for the biological response because it affects the ability of particles to cross biological barriers. The most common shapes are rods, spheres, cylinders, and cubes. Spherical shapes are reported to have less toxicity than star-shaped counterparts on human skin fibroblasts. Branched-shaped nanoparticles were reported to have high toxicity due to their ability to cling to cells for longer periods of time, causing more damage [
54,
55,
56,
57,
58,
59].
Polymeric nanoparticles can present a nanocapsule or a nanosphere morphology. Nanocapsules have an oily core surrounded by a thin polymeric envelope. Nanospheres are made of a solid polymeric network. A PS or a drug can be dissolved in the core or adsorbed to the polymeric wall in polymeric nanoparticles. The drug release should be controlled temporal and spatially to reduce side effects and prolong therapeutic effects as desired. Drug diffusion can be affected by the presence of water because the pores of the polymeric matrix become enlarged, allowing drug diffusion. Additionally, the thickness of the polymeric envelope and the permeability of the drug impact its release through the polymeric matrix. The degradation rate of the polymer is also very important [
60,
61,
62,
63]. Polymeric nanoparticles size usually ranges from 100 to 300 nm, but dimensions under 100 nm have also been reported. The polymers used can have a crystalline or amorphous structure, which will impact the physicochemical properties of the nanoparticles, including their drug release performance [
64,
65].
Lipid-based nanoparticles can be liposomes that have a core-shell nanostructure making them appropriate for loading hydrophobic and hydrophilic molecules. Hydrophobic molecules are encapsulated in the lipophilic bilayers of the shell, and hydrophilic molecules are entrapped in their core. They can also be solid lipid nanoparticles (SLN), composed of a solid lipid core stabilized in aqueous media by surfactants. SLN usually present improved stability, protection, and controlled release. However, the drug loading capacity is limited by their polymorphism. The nanostructured lipid carriers (NLC) are composed of a mixture of liquid and solid lipids, with a liquid lipid range between 10 and 30%. NLCs have improved drug retention and enhanced drug loading capacity and are divided into: (i) imperfect type with a highly disordered matrix leading to high drug loading capacity but low encapsulation efficiency; (ii) amorphous type, that contains a structureless solid matrix to avoid crystallization-induced drug leakage; (iii) multiple type with oil nanocompartments distributed in the solid matrix that acts as a barrier preventing drug leakage and controlling drug release. The drug solubility in an oil nanocompartment is higher than in a solid matrix enabling higher drug loading [
66,
67,
68,
69,
70,
71]. Their size range between 10 and 1000 nm, but there are also lipid-based nanoparticles between 10 and 150 nm, the ideal ones for drug delivery since they can transport hydrophobic and hydrophilic molecules, can be surface modified and offer controlled drug release [
66].
Two-dimensional nanomaterials have a sheet-like structure and lateral dimensions larger than 100 nm but a thickness usually below 5 nm. They have a large surface area and anisotropic physical and chemical properties. Two-dimensional nanomaterials can be, for example, carbon-based materials (e.g., graphene, graphene oxide), clay materials, transition metal oxides or black phosphorus. These materials can be layered: the layers stack together due to the weak Van der Waals interaction, and inside them, strong chemical bonds connect the atoms. The arrangement of the atoms determines the physicochemical properties. Two approaches may be applied to prepare 2D layered nanomaterials: (i) the top-down approach uses driving forces to break the van der Waals interactions between layers and achieve exfoliation into 2D primary sheets; (ii) the bottom-up one relies on assembly of 2D nanosheets following an appropriate chemical synthesis route [
72,
73,
74,
75,
76,
77].
2.2. Surface Modification and Encapsulation Strategies
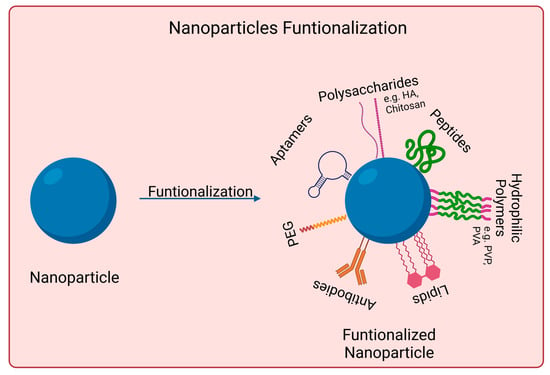
Nanomaterials to be used for biomedical applications should have good stability in physiological conditions and biocompatibility. Many nanoparticles agglomerate in aqueous solutions due to being hydrophobic or creating strong interparticle interactions, for instance. Covalent and non-covalent surface functionalization can be performed to enhance nanoparticles properties. However, such modifications should not affect nanomaterials photoabsorption properties if skin cancer photoimmunotherapy applications are desired. Figure 2 shows examples of such approaches.
Figure 2. Surface functionalization strategies used for skin cancer photoimmunotherapy. Functionalization with PEG (polyethylene glycol), polysaccharides, lipids, polymers, aptamers, peptides, and antibodies. Created in Biorender.com. Abbreviations: HA, hyaluronic acid; PVP, poly-(vinylpyrrolidone); PVA, Polyvinyl alcohol. Created with BioRender.com.
Functionalization with PEG usually increases the nanoparticles biocompatibility and blood circulation time. It also decreases the interaction with the targeted cells [
78,
79,
80,
81,
82]. It is relevant to mention that functionalization often changes nanomaterials physicochemical properties, such as conformation, electrostatic binding, and hydrophobicity, which might impact not only its biointeractions but also its photo/immunotherapy effectiveness. There are already approved PEGylated drugs by FDA, such as Adagen
® and Oncaspar
® [
83]. Other synthetic polymers used for surface coating include polyethyleneimine (PEI), polyvinyl alcohol (PVA), and polyvinylpyrrolidone (PVP). Some other surface modifications strategies include the use of polysaccharides, namely, chitosan or hyaluronic acid. These act as steric protection against protein adsorption and macrophage uptake. They are also biocompatible and biodegradable. When coating metallic nanoparticles, they can also help prevent oxidation and release of metal ions [
78,
79,
80,
81,
82].
Antibodies possess high target specificity and affinity for certain molecules at the cell surface. They can be used, for example, to direct particles for selective drug delivery or phototherapy. Furthermore, the potential high recognition capabilities of antibody-modified particles might be able to reduce immunological side effects [
78,
79,
80,
82,
84]. Aptamers are synthetically prepared short oligonucleotides. They can interact with cellular targets (e.g., nucleic acids, transmembrane proteins, sugars) with high affinity and selectivity. They present advantages over antibodies, such as having a smaller size, a higher ratio of target accumulation, and higher in vivo stability. However, their half-time circulation is shorter [
79,
80,
82,
85]. Short peptides have also been used for surface functionalization because they offer several advantages, such as increased stability, reduced immunogenicity, and versatile methods for conjugation with nanoparticles. Small molecules can recognize some markers or receptors on the surface of target cells, increasing nanoparticle uptake [
79,
80,
82].
Lipids are another surface modification strategy. Lipid-coated nanoparticles present several advantages, such as reduced cytotoxicity and better target specificity for drug delivery. Natural phospholipids are often used to coat polymeric nanoparticles. Their amphiphilic nature allows the formation of membrane-mimetic structures on nanoparticles [
79,
80,
82,
86].
Encapsulation is the method of enclosing molecules inside a shell or carrier, which can be polymers or lipids. It can be used to protect the molecules from degradation, preserve their properties as well as control their release in the body. Encapsulation can help to deliver the molecules effectively, at a controlled rate, helping to reduce the potential side effects. There are several encapsulation techniques, and the appropriate choice depends on the specific molecule, such as its size, solubility, surface charge, and the desired therapeutic effect. Examples of encapsulation techniques to produce polymer-based nanoparticles are nanoprecipitation or emulsion evaporation; for liposomes, production techniques such as hydration of a thin film or solvent injection can be used [
87,
88,
89]. Nanoprecipitation is a simple technique where non-water-soluble polymer and drug(s) are dissolved in the same organic solvent. The resultant dispersion is added to an aqueous solution that contains a surfactant under stirring, and the solvent diffuses into the aqueous phase, causing the polymer to precipitate and form the nanoparticles [
88,
90]. The emulsion evaporation method consists in dissolving the polymer and drug in a water-immiscible organic solvent that is later added to an aqueous phase containing a suitable stabilizer. Then, the organic solvent is usually evaporated under stirring to obtain the nanoparticles. High-shear mixing can be used to decrease particle size to the nanometric scale and to narrow size distribution [
88,
89]. The hydration of a thin film technique involves dissolving the lipids in an organic solvent; the solvent is then evaporated to form a thin lipid film. Finally, hydration with water or a buffer is performed to enable the formation of a liposome dispersion. Sonication is usually performed to increase uniformity and reduce particle size. The solvent injection technique consists of dissolving phospholipids in an organic solvent (e.g., ethanol, ether) that is then injected into an aqueous buffer solution. Lipids self-assembly forming liposomes following ethanol dilution or ether evaporation. This method presents the advantage of allowing to obtain of small nanoscale liposomes (usually below 100 nm) with a narrow size distribution, without extrusion or sonication [
88].
3. Skin Cancer Photoimmunotherapy Studies
Skin cancer is one of the most common cancers. Although there are already several treatment options available, there is still an urgent need to reduce mortality, recurrence rates, and side effects [
3,
8,
91]. Photoimmunotherapy has gained much interest in recent years. It combines the advantages of phototherapy with enhancement of the immune response, resulting in a more effective cancer treatment approach.
Figure 3 shows the mechanisms of action of photoimmunotherapy.
Table 1 summarizes the state-of-the-art literature regarding nanomaterials used for skin cancer photoimmunotherapy, including their composition, size, and biological effects.
Figure 3. Skin cancer photoimmunotherapy using functionalized nanoparticles. The nanoparticles can exert a direct effect on cells by destroying primary tumors and activating immune cells. Photoimmunotherapy can induce an inflammatory response and increase the release of pro-inflammatory cytokines (e.g., TNF-α, IFN-γ, IL-6, IL-12). Nanoparticles were modified using specific drugs, polymers, or antibodies to induce a desired immune response. Abbreviations: TSAs, tumor-specific antigens; DAMPs, damage-associated molecular patterns; IL-6, interleukin-6; IL-12, interleukin-12; TNF-α, tumor necrosis factor alpha; INF-γ, interferon gamma. Created with BioRender.com.
Table 1. Nanomaterials for skin cancer photoimmunotherapy. Their properties, composition, and biological effects.
|
Nanomaterial
|
Physicochemical Modifications
|
Loaded
Substance
|
Particle Size
|
In Vitro Studies
|
In Vivo Studies
|
Ref.
|
|
Parameters
|
Results
|
Parameters
|
Results
|
|
Aluminum hydroxide
|
BSA surface adsorption
|
Chlorin e6
|
25.25 ± 2.1 nm
|
B16F10 cells
[Al-BSA-Ce6] = 0.1 μg mL−1
I: 660 nm, 0.8 W cm−2, 5 min
|
95% cell death (MTT assay)
↑CD80
↑TNF-α, IL-12p70, IL-1β
|
C57BL/6 mice subcutaneously injected with B16F10 cells
[Al-BSA-Ce6] = 5 mg kg−1
I: 660 nm, 0.8 W cm−2, 5 min
|
Tumor volume of 0 mm3 at day 7
≈63% mice survived 100 days
↑T cells tumor infiltration
↑TNF-α and INF-γ production
|
[92]
|
|
Black Phosphorus
|
PEG electrostatic adsorption
|
Imiquimod
|
≈120 nm
|
B16 cells
[BP-PEG] = 10 μg mL−1
I: 808 nm, 3.2 W cm−2, 10 min
|
45% cell viability↓ (MTT assay)
↑TNF-α, IL-6, IL-12
DCs↑ 30.8%
|
C57BL/6 mice subcutaneously injected with B16
[BP] = 0.5 mg kg−1, 25 μL
[R837] = 0.35 mg kg−1, 25 μL
I: 808 nm, 3.2 W cm−2, 3 min
|
≈10-fold tumor vol.↓
DCs↑ 45.5%
↑TNF-α, IL-6, IL-12
|
[93]
|
|
Black Phosphorous
|
N/A
|
FKF-OVAp
|
≈500 × 23 nm
|
N/A
|
N/A
|
C57BL/6 mice subcutaneously injected with B16-OVA
[FKF-OVAp] = 10 nmol per mouse
[BPs] = 15.9 μg per mouse
I: 808 nm, 0.5 W cm−2, 5 min
|
≈3-fold tumor vol.↓
100% survival over 60 days
↑DC activation
↑CD8+ T cells effector and central memory
|
[94]
|
|
Chitosan
|
Cross-linking, sodium tripolyphosphate
|
IDO
|
220 nm
|
B16 cells
[ICG-NP] = 30 μg mL−1
I: 808 nm, 0.35 W cm−2, 5 min
|
≈0% cell viability
(CCK8 assay)
≈85% DC frequency
|
C57BL/6 mice subcutaneously injected with B16F10
Drug loading: 4 and 35 μg per microneedle patch of ICG and 1-MT, respectively
I: 808 nm, 0.35 W cm−2, 5 min
3 cycles at interval of 2 days
|
Tumor volume of ≈0 mm3
80% survival rate without recurrence after 120 days
≈55% DC maturation level
↑CD8+ T cells in distant tumor
↑TNF-α, IL-12p70, IL-6
|
[95]
|
|
Gold
|
HA surface adsorption
|
M2pep
|
64.6 nm
|
B16F10 cells
[HA-AuNR/M-M2pep] = 20 μg mL−1
I: 808 nm, 1.5 W cm−2, 2 min
|
40% cell viability
(CCK8 assay)
35.1 ± 1.8% apoptosis
(Annecin V-FITC)
↑CRT-positive cells and HMGB1 release
|
C57BL/6 mice subcutaneously injected with B16F10
[AuNR] + [M2pep] = 10 + 12 mg kg−1
I: 808 nm, 1.5 W cm−2, 2 min
|
≈10-fold tumor vol.↓
67% survival rate at 45 days
3.7-fold↑ CD8+ T cells
INF-γ, TNF-α↑ ≈ 6-fold
|
[96]
|
|
Gold
|
BSA surface adsorption
|
R837
|
122.1 ± 11.6 nm
|
B16-F10 cells
[Au] = 11.5 μg mL−1
I: 1064 nm, 1.0 W cm−2, 10 min
|
Cell viability↓ to ≈27%
(MTS assay)
HSP70/β-Actin release ≈ 0
|
C57BL/6 mice subcutaneously injected with B16F10
[Au] = 300 μg mL−1
I: 1064 nm, 1.0 W cm−2, 10 min
|
≈10-fold tumor vol.↓
TNF-α, IL-6, IL-12 ≈ 14, 9, 3 times↑ than PBS, respectively
↑CD8+ T cells infiltration
|
[97]
|
|
Gold
|
Gold nanoparticles retained in extracellular vesicles with tumor antigens (AuNP@B16F10)
|
Tumor antigens (AuNP@B16F10)
|
40 nm
|
N/A
|
N/A
|
Murine melanoma model subcutaneously injected with AuNP@DCB16F10
[Au] = 1.35 mg kg−1
I: 808 nm, 2.0 W cm−2, 1 min
Cycle: 3 times, 3 days interval
|
69% tumor volume↓
50% tumor-free mice at day 19
Distant tumor inhibition
↑CD3+ and CD8+ T cells infiltration
↑INF-γ, TNF-α, IL-6
|
[98]
|
|
Gold
|
N/A
|
SV
|
50 nm
|
B16-F10 cells
[Au] = 60 μg mL−1
I: 808 nm, 1.5 W cm−2, 2 min
|
≈12% cell viability
(MTT assay)
CRT expression
≈3.5-fold↑
↑mature DCs frequency
|
B16F10-bearing C57 mice
I: 808 nm, 1.5 W cm−2, 2 min
|
≈10-fold tumor vol.↓
↑DC maturation
CD4+ and CD8+ T cells proliferation
|
[99]
|
|
Hyaluronic acid
|
Self-assembly of Ce6/α-linoleic acid (L-Ce6 NAs (nano-assemblies))
Fast dissolving L-Ce6 NAs in oligo-HA and micro-molding of microneedles (tips enriched with 3 µg Ce6)
|
Ce6
|
≈86 nm
|
B16F10 cells
[Ce6] = 400 μM
I: 660 nm, 200 mW cm−2, 5 min
|
CRT fluorescence ↑2-fold
ATP secretion ≈ 2.5 nM
↑HMGB1 release
|
C57BL/6 mice subcutaneously injected with B16F10
[Ce6] = 0.12 mg kg−1
I: 660 nm, 200 mW cm−2, 4 min
|
≈3-fold tumor vol.↓
↑CD4+ and CD8+
≈3 and 4-fold
|
[100]
|
|
Liposomes
|
N/A
|
TRP-2
|
180.4 ± 10.2 nm
|
B16F10 cells
[TLipIT NPs] = 100 μg mL−1
I: 808 nm, 0.75 W cm−2, 5 min
|
≈12% early apoptosis
(Annexin V-FITC)
37% late apoptosis
↑TNF-α, INF-γ
|
C57BL/6 mice subcutaneously injected with B16F10
[TLipIT/NEs] = 100 μg mL−1
I: 808 nm, 0.75 W cm−2, 5 min
|
≈10-fold tumor vol.↓
≈33% CD80+ and CD86+ mature DCs frequency
≈49 and ≈33%
CD4+ and CD8+ T lymphocytes frequency
|
[101]
|
|
Micelles
|
N/A
|
CQ
IR780
|
80−90 nm
|
B16 cells
[C/I-Mil] = 4 μg mL−1
I: 808 nm, 1.0 W cm−2, 5 min
|
20% cell viability
(CCK-8 assay)
Cell membrane integrity destroyed
Phagocytic index
↑3.0-fold
|
C57BL/6 mice subcutaneously injected with B16F10
[C/I-Mil] + [CQ/Mil] = 4 μg mL−1 + 20 μg/patch
I: 808 nm, 1.0 W cm−2, 5 min
|
Primary tumor suppression: 0 mm3
50% survived at least 40 days
Distant tumor volume ↓3.4-fold
|
[102]
|
|
Micelles
|
N/A
|
Imiquimod
|
72.0 ± 18.0 nm
|
N/A
|
N/A
|
C57BL mice subcutaneously injected with
B16 cells
[IQPM] = 5 mg kg−1
I: 808 nm, 1.5 W cm−1, 5 min
|
Primary tumor suppression: 0 mm3
CD8+ and CD4+ T cells ↑9.3- and 10.3-fold
2.4-fold↓ metastasis
|
[103]
|
|
mPEG-Pep-IDOi/ICG NPs
|
N/A
|
N/A
|
140 nm
|
B16-F10 cells
[ICG] = 20 μg mL−1
I: 808 nm, 1.0 W cm−2, 5 min
|
≈0% cell viability
(CCK-8 assay)
Induced ICD of tumor cells
≈70% CD80 and CD86↑
|
C57BL/6 mice subcutaneously injected with B16-F10
[ICG] = 4 mg kg−1
[IDOi] = 5 mg kg−1
I: 808 nm, 1.0 W cm−2, 5 min
|
Primary tumor suppression: 0 mm3
CD80+ and CD86+
↑13.5 and 12.3%
↑INF-γ, TNF-α, IL-6
|
[104]
|
|
PBE
|
N/A
|
RSL-3
|
<100 nm
|
B16-F10 cells
[RSL-3] = 0.5 μg mL−1
[INF-γ] = 100 ng mL−1
I: 671 nm, 100 mW cm−2, 1 min
|
30% mature DC
CTR expression
↑5.0-fold
|
C57BL/6 mice subcutaneously injected with B16-F10
[RSL-3] = 0.5 μg mL−1
[INF-γ] = 100 ng mL−1
I: 671 nm, 150 mW cm−2, 2 min
|
≈2-fold tumor vol.↓
≈50% survival rate
≈30% mature DC cells
INF-γ secretion ↑ 4-fold
|
[105]
|
|
PEI-PBA
|
PEG surface adsorption
|
Ce6
aPDL1
|
117 ± 4.0 nm
|
B16F10 cells
[NC@Ce6-pH 6.0] = 7.5 μg mL−1
I: 650 nm, 20 mW cm−2, 2.5 min
|
89% CRT rate
≈34% apoptosis
(Annexin V-FITC)
DC maturation
|
B16F10 tumor-bearing mice
[Ce6] = 2.5 mg kg−1
I: 650 nm, 100 mW cm−2, 10 min
|
78% tumor inhibition rate
≈49% tumor infiltrating T cells
DC maturation
|
[106]
|
|
Polydopamine
|
PEI surface adsorption
|
CpG oligodeoxynucleotides
|
140 nm
|
B16F10 cells
[PPP/CpG/HA] = 200 μg mL−1
I: 808 nm, 2.0 W cm−2, 5 min
|
≈5% cell viability
(MTT assay)
≈60% apoptosis
(Annexin V-FITC)
≈60% CD80+ DC
≈50% CD86+ DC
|
C57BL/6 mice subcutaneously injected with B16F10
[PPP/CpG/HA] = 0.75 mg kg−1
I: 808 nm, 1.5 W cm−2, 5 min
|
≈20-fold tumor vol.↓
Largest apoptotic cell area
CD80+ DC ≈ 3%
CD86+ DC ≈ 4.5%
|
[107]
|
|
Silicon Dioxide
|
CuS loaded inside the pores
PDMAEMA surface adsorption
|
IL-12 gene
|
157 nm
|
B16F10 cells
[CSP] = 34.5 μg mL−1
I: 1064 nm, 0.65 W cm−2, 5 min
|
<20% cell viability
(CCK-8 assay)
89% apoptotic cells
(Annexin V-FITC)
↑CRT expression
66% DCs maturation
|
B16F10-bearing C57BL/6 mice
[CSP] = 172.4 μg per mouse
I: 1064 nm, 0.65 W cm−2, 5 min
|
≈3-fold tumor vol.↓
Prolonged survival
DC maturation level: ≈48%
21% CD4+ and 12% CD8+ T populations
|
[108]
|
|
Silicon Dioxide
|
Chemical synthesis of UCNP@m-SiO2@liposome NPs
|
Ce6 and BSO
|
≈50 nm
|
B16/F10 cells
[UCB] = 100 μg mL−1
I: 980 nm, 0.7 W cm−2, 10 min
|
≈29% cell viability
(CCK-8 assay)
≈39% apoptosis rate
(Western Blot)
↑TNF-α, IL-6, INF-γ
|
C57BL/6 mice subcutaneously injected with B16F10
[UCB] = 0.8 mg per mouse
I: 980 nm, 0.7 W cm−2, 20 min
|
≈3-fold tumor vol.↓
↑IL-12p40, INF-γ
|
[109]
|
Abbreviations: ↑, increase; ↓, decrease; aPDL1, anti-programmed death-ligand 1; BSA, bovine serum albumin; B16 cells, B16 murine melanoma cell line; BSO, buthionine sulfoximine B16F10 cells, B16F10 murine melanoma cell line; B16-F10 cells, B16–F10 murine melanoma cell line; B16/F10 cells, B16/F10 murine melanoma cell line; CCK-8, cell counting kit-8; Ce6, chlorin e6; CQ, chloroquine; DC, dendritic cells; HA, hyaluronic acid; IDO, indoleamine 2,3-dioxygenase; M2pep, M2 macrophage-targeting peptide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; N/A, not applicable; PDMAEMA, poly((2-di- methylamino)ethyl methacrylate); PEG, polyethylene glycol; PEI, polyethyleneimine; R837, imiquimod; SV, simvastatin; TRP-2, tyrosinase-related protein 2.