Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Patients with inflammatory bowel disease (IBD) are at an increased risk of developing colorectal cancer (CRC), mainly because of chronic intestinal inflammation. Some unique molecular differences occur in colitis-associated CRC, resulting in a different sequence of events, primarily of inflammation–dysplasia–carcinoma, compared to sporadic cases.
- carcinogenesis
- colitis-associated cancer
- inflammation
- IBD therapy
- gut barrier
- OMIC
- microbiome
1. Introduction
Inflammatory bowel disease (IBD), comprising ulcerative colitis (UC) and Crohn’s disease (CD), are chronic relapsing inflammatory disorders in which a complex interplay of genetic predisposition and environmental factors alter host–microbiota interactions and lead to dysregulation of gut immune responses in a genetically susceptible individual [1]. The literature has widely consolidated that IBD is associated with an increased risk of colorectal cancer (CRC) compared to the general population [2][3]. A meta-analysis of 116 studies by Eaden et al. [3] reported that the incidence of IBD-associated CRC was 2%, 8%, and 18% at 10, 20, and 30 years after the onset of UC, respectively. Concerning CD, the risk of CRC is debatable and considered slightly elevated compared to the general population. A subsequent meta-analysis [4] estimated a cumulative risk of 2.9% at 10 years, 5.6% at 20 years, and 8.3% at 30 years in patients with CD. Several risk factors are associated with the development of colonic neoplasia, including longer disease duration, greater extent of colonic involvement, a family history of CRC, primary sclerosing cholangitis, male sex, and younger age at diagnosis. There is also additional risk related to worse disease severity, including high inflammatory burden, backwash ileitis, pseudopolyps, prior dysplasia, and colonic strictures [2]. However, recent evidence suggests that the incidence rates of IBD-associated CRC decreased over the last decade due to better control of inflammation and advances in the endoscopic detection and resection of precancerous lesions which have improved surveillance programs [5]. Nevertheless, it should be noted that most of these data came from tertiary referral centres wherein the study population was selected.
IBD-associated CRC (IBD–CRC) has been considered a distinct entity compared to sporadic CRC (sCRC), and little is known about the pathogenesis and mechanisms behind the IBD–CRC [6]. The key driver of neoplastic changes and progression is chronic inflammation that contributes to dysplasia and is considered the most critical risk factor for developing colitis-associated CRC [7].
Chronic inflammation generates oxidative stress-induced DNA damage that may activate tumour-promoting genes and inactivate tumour-suppressing genes [8]. As a result, markers of oxidative damage and DNA double-strand breaks increase progressively in the inflammation–dysplasia–carcinoma sequence rather than the ‘adenoma–carcinoma’ sequence typical of sCRC (Figure 1). In addition, recent studies have demonstrated a potential role for the gut microbiome and host immune system. These lead to subsequent events that cause genetic and epigenetic alterations followed by clonal expansion of somatic epithelial cells, influenced by surrounding stromal and immune cells [9].
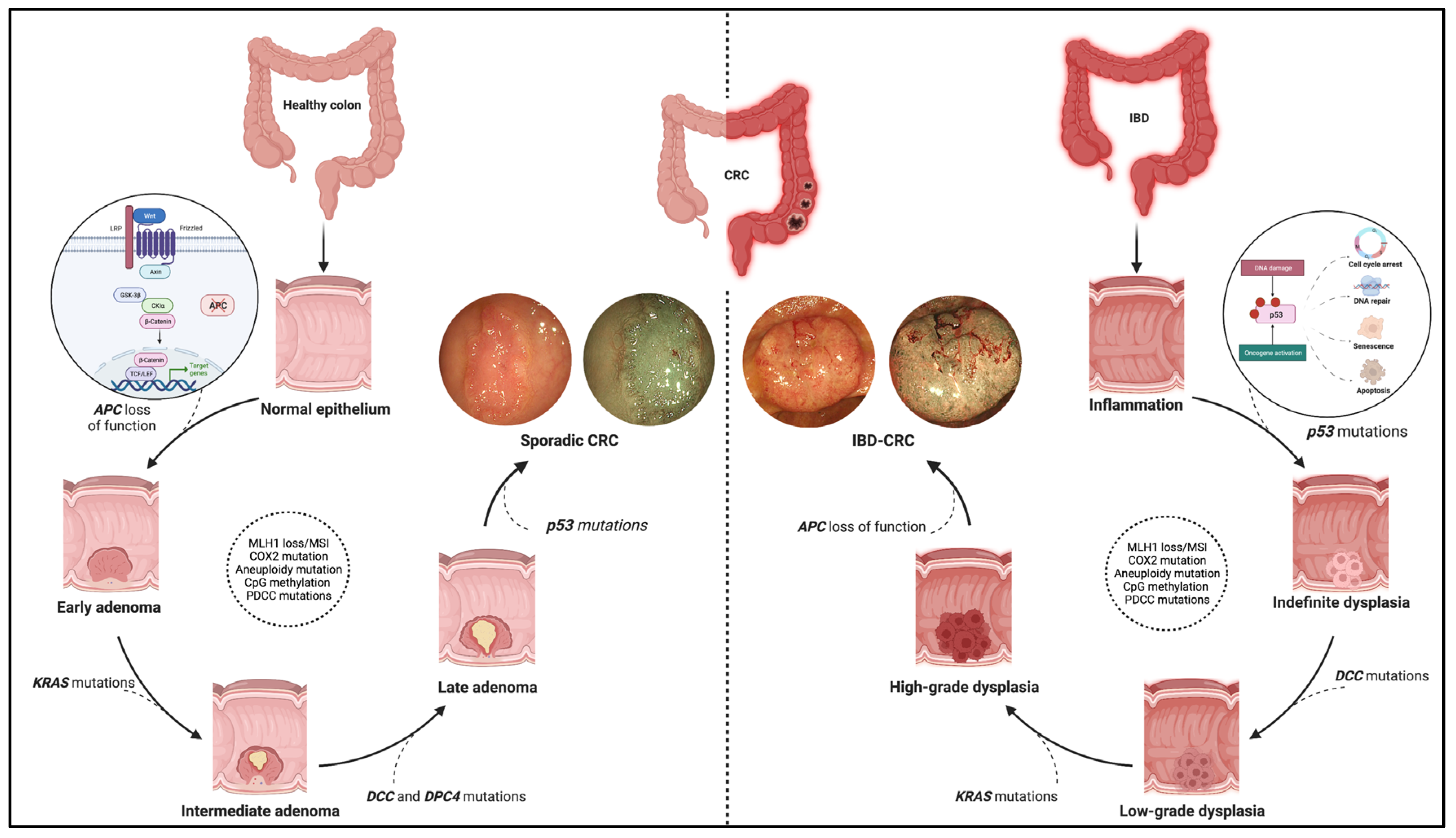
Figure 1. Pathogenic pathway of sporadic colorectal cancer and IBD-related colorectal cancer. The figure shows the different molecular pathways related to sporadic and IBD-related colorectal cancer (CRC). The sporadic CRC prevails in the adenoma-to-carcinoma sequence, while the inflammation–dysplasia–carcinoma cascade characterises the colitis related-CRC. In addition, the main gene mutations determining the progression of the two tumour phenotypes are reported with emphasis on the earliest mutations in the two processes. Namely, these entail APC loss of function, the WNT-beta catenin pathway activation for sporadic CRC (see zoom circle at left), and p53 mutations with consequent impacts on cell cycle, DNA repair, and cell viability for IBD-related CRC (see zoom circle at right). Finally, high-definition white light endoscopic images and virtual chromoendoscopic images (obtained through the Narrow Band Imaging technology) are provided for each phenotype of tumours. Created with “Biorender.com”. Abbreviations: CRC, colorectal cancer; IBD, inflammatory bowel disease.
2. IBD–CRC: A Distinct Molecular Pathway
The molecular pathways occurring in IBD–CRC differ from those observed in sCRC. The adenoma-to-carcinoma pathway prevails in sporadic cancer, while the sequence inflammation–dysplasia–carcinoma characterises the colitis related-CRC (Figure 1).
More in-depth, constitutive activation of WNT/b-catenin signalling via loss-of-function mutations in APC is the primary and earliest player of sCRC development, regulating cell fate, proliferation, polarity, and stemness via b-catenin-dependent and independent mechanisms [10]. Multiple other driver genes, such as KRAS, P53, PIK3CA, SMAD4, ARID1A, MYC, etc., are involved in the following progression of sCRC. These genes are also involved in IBD–CRC even though the timing and frequency of some of the common gene alterations are different [11][12]. Notably, mutation and loss of function occur frequently and very early in the process, even before dysplasia, suggesting an alternative mechanism of WNT pathway activation [6]. Moreover, KRAS mutations occur less frequently and later in IBD–CRCs; nuclear accumulation of b-catenin is prevalent compared to sCRCs.
Interestingly, a “Big Bang” model of CRC evolution has been formulated to support this concept. Chronic inflammation induces tumour-promoting molecular alterations in pre-existing clonal cell populations that occur sharply rather than a gradual accumulation driven by pressures from the microenvironment [6][13].
A powerful tool to describe tumour transcriptional, genetic, epigenetic, and microenvironment characteristics is a transcriptome-based classification of CRCs that consists of four consensus molecular subtypes (CMSs).
CMS1 (microsatellite instability immune, overall 14%) characterised by hypermutated, microsatellite unstable and strong immune activation; CMS2 (canonical, overall 37%) marked by epithelial cell differentiation with upregulated of WNT and MYC signalling activation; CMS3 (metabolic, overall 13%), with epithelial and evident metabolic dysregulation; and CMS4 (mesenchymal, overall 23%) mediate the activation of the pathway related to the epithelial–mesenchymal transition and stemness with transforming growth factor–β activation and a prominent stromal invasion [11][14].
Regarding IBD–CRC, the CMS distribution has yet to be fully clarified even though a complete lack of CMS2 tumours and a skewing towards the CMS4-associated epithelial–mesenchymal transition pathway is the most prevalent. These are associated with EMT, matrix remodelling, transforming growth factor b (TGF-b) signalling, complement activation, and depletion of WNT/MYC-related expression signatures [6].
Rajamäki K et al. [11] have recently analysed whole-genome sequencing, single nucleotide polymorphism arrays, RNA sequencing, genome-wide methylation analysis, and immunohistochemistry from 31 patients with IBD–CRC.
Notably, they reported a complete absence of canonical epithelial tumour subtypes associated with WNT signalling CMS2 tumours and a predominance of the mesenchymal tumours related to oncostatin M receptor overexpression CMS4 among IBD–CRCs
Additionally, the propensity for a relative loss in expression of HNF4, a transcription factor essential for the embryonic development of colonic epithelium and the overexpression of a receptor for cytokine oncostatin M (OSMR), which may contribute to a more mesenchymal phenotype was observed.
Elevated intestinal OSMR and OSM expression are also associated with a subgroup of IBD patients showing poor response to TNFa blockers. Hence, OSM could be considered a potential biomarker and therapeutic target for IBD. However, further studies are required to establish the non-invasive detection of colorectal adenomas and carcinomas from biomarkers.
3. OMICS: Future Directions
Multi-omics studies consisting of meta transcriptomics, metaproteomic, and metabolomics offer a great chance to fill knowledge gaps in IBD pathogenesis for a better understanding of CRC development as well as its classification into different molecular subtypes for patient stratification and the development of new biomarkers and targeted therapies [15] (Figure 2).
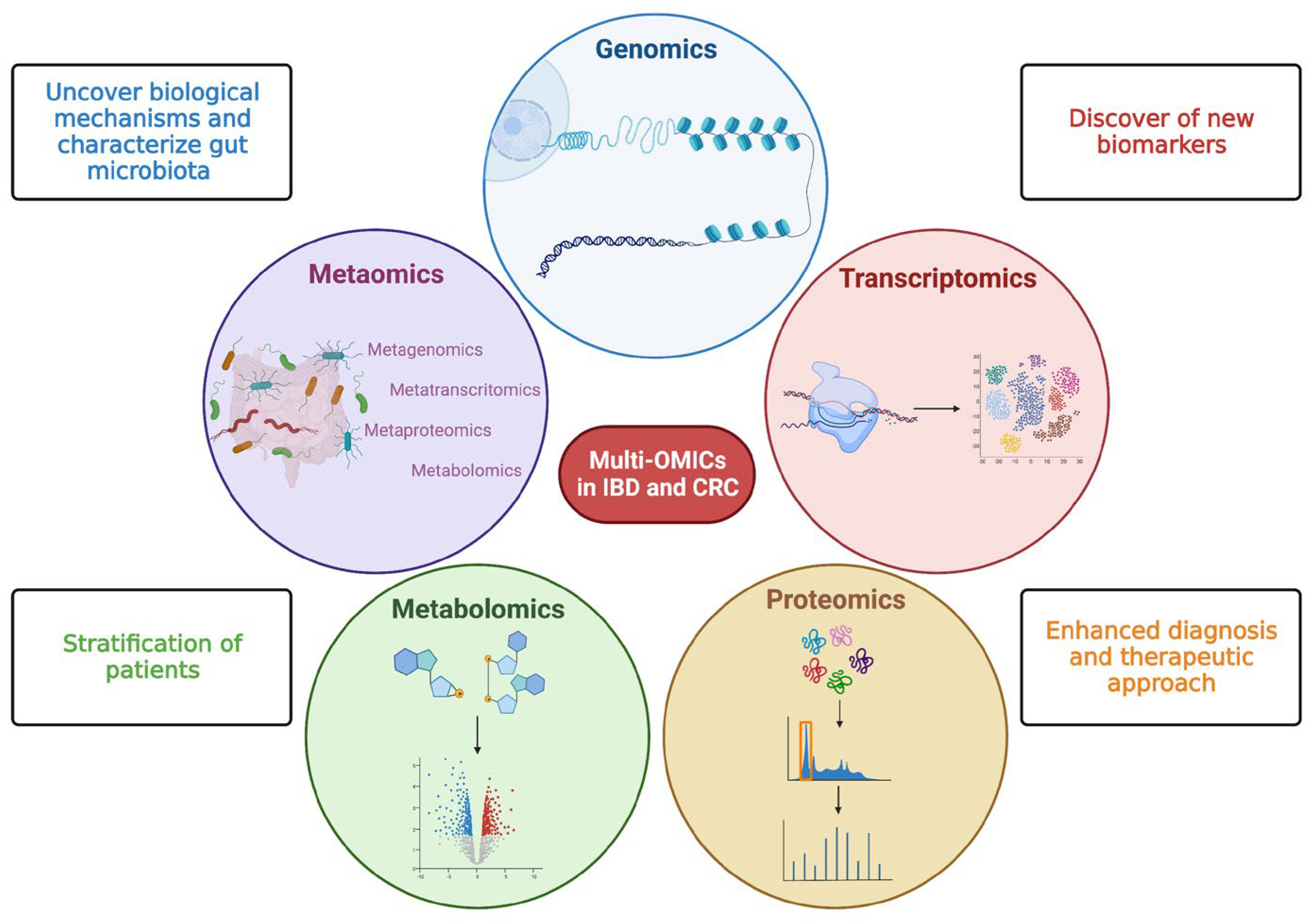
Figure 2. Multi—OMICs and its impact on inflammatory bowel disease and colorectal cancer. This figure schematically represents the main OMIC techniques available to date: genomics—the study of the genetic or epigenetic sequence information; transcriptomics—the evaluation of RNA transcripts; proteomics—the investigation of the structure and function of proteins; metabolomics—the identification and quantification of metabolites; metaomics—the application of the previously described techniques to the gut microbiome. The multiple and integrated application of these techniques, so-called multi-OMICs, will offer a great chance to fill knowledge gaps in inflammatory bowel disease (IBD) and colorectal cancer (CRC). In more detail, the application of multi-OMICs will provide (as specified in figure squares) the discovery of novel biological mechanisms below IBD and CRC pathogenesis, the detection of new clinically relevant biomarkers, the definition of integrated signatures able to stratify patients, and the enhancement of physician ability to establish disease prediction, establish a prognosis, and treat patients appropriately. Created with “Biorender.com”. Abbreviations: CRC, colorectal cancer; IBD, inflammatory bowel disease.
Further OMICs approaches allow better characterisation of the gut microbiome and how the microbiome in turn influences the surrounding ecosystem in order to identify why some IBD patients develop CRC and others do not [16].
However, most studies are based on individuals with sporadic CRC, whilst there is still a relatively small number of studies integrating omic datasets from IBD patients. The integrative human microbiome project (iHMP) collects host and microbiome-associated data using multiple meta-omics strategies [17][18]. Specifically, this project aims to identify microbial community changes over time that precedes human diseases such as IBD [19].
More recently, a comprehensive multi-omics project on the pathogenesis and outcomes of primary sclerosing cholangitis (PSC) associated with IBD was developed. Based on the hypothesis that multi-omics analyses of data capturing environmental exposures and the associated biological responses, the project aims to better understand the role and interplay of the genome, exposome, and microbiome on pathways influencing PSC pathogenesis and outcomes [20].
4. Primary Sclerosing Cholangitis (PSC) as a Risk Factor for IBD–CRC
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease with a pooled prevalence in patients with IBD of 2.16%. When low-grade dysplasia is detected in patients with PSC–IBD, they are at higher risk of developing advanced CRC compared to patients with IBD without PSC, suggesting a more aggressive disease course [2].
Hence, in IBD with concomitant primary sclerosing cholangitis (PSC) the risk of CRC increases substantially, yielding a 3-to 4-fold higher risk compared with having IBD alone [21]. In an attempt to explain this augmented risk, in a recent study, de Krijger M et al. [22] analysed DNA copy number variations, microsatellite instability (MSI), mutations on 48 cancer genes, and the CpG island methylator phenotype (CIMP) from resection specimens of 19 patients with PSC–IBD–CRC. Furthermore, they compared these genetic profiles with two published cohorts of IBD-associated CRC (IBD–CRC; n = 11) and sporadic CRC (s-CRC; n = 100). The excess risk of CRC in patients with PSC–IBD could not be explained by copy number aberrations, mutations, MSI, or CIMP status in cancer tissue or in adjacent mucosa.
Most importantly, no significant differences were found in patterns of chromosomal aberrations in PSC–IBD–CRC concerning those observed in IBD–CRC and s-CRC. Mutation frequencies were similar between the groups, except for mutations in KRAS, which were less frequent in PSC–IBD–CRC (5%) versus IBD–CRC (38%) and sCRC (31%; p = 0.034) and in APC which were less frequent in PSC–IBD–CRC (5%) and IBD–CRC (0%) versus sCRC (50%; p < 0.001). Notable cases of PSC–IBD–CRC were frequently CIMP positive (44%), similar to sCRC (34%; p = 0.574), whilst they were less frequent than in patients with IBD–CRC (90%; p = 0.037) suggesting that epigenetic changes may play an important role. Hence, these findings pave the way for further exome-wide and epigenetic studies.
5. The Role of the Intestinal Barrier in Colorectal Cancer Development
The interplay between the intestinal barrier and microbiome is crucial in IBD pathogenesis. The impairment of the intestinal barrier (especially of the epithelium) and a concomitant microbiome alteration (defined as ‘dysbiosis’) are considered the cornerstones of disease development [23][24]. While it is unclear which is the primum movens in this process, what is certain is that the alteration of mucosal barrier integrity, with the impairment of tight junctions (TJ) and the interaction with a harmful microbiome act as triggers for immune processes in the lamina propria, activating inflammatory immune processes, with possible cancer development (Figure 3).
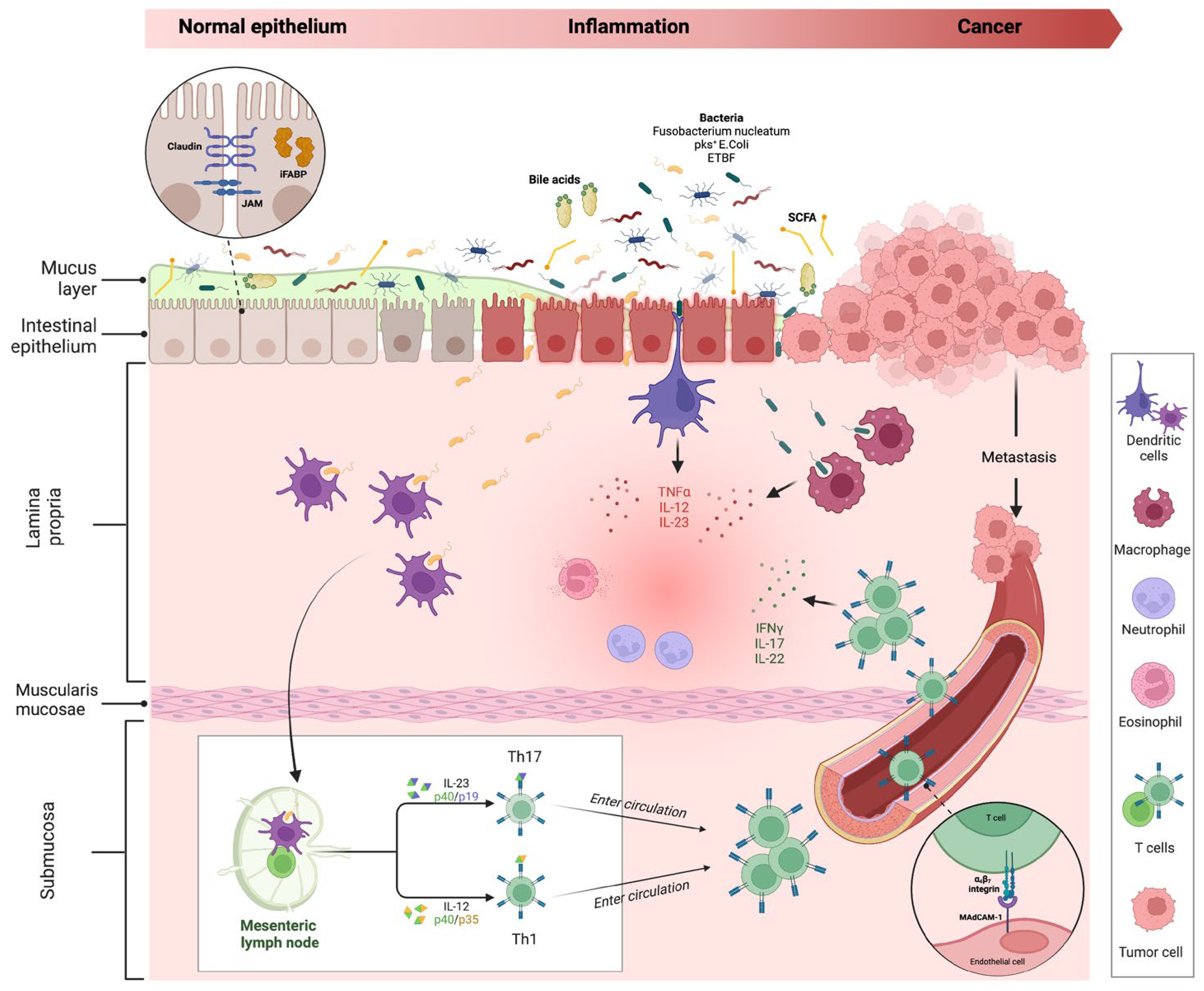
Figure 3. Intestinal inflammation and evolution to cancer in IBD. The impairment of the mucus layer and epithelial barrier, associated with dysbiosis, determines the inflammatory response, leading to IBD disease development and possible cancer evolution. Bile acids and small-chain fatty acids participate in initiating this process. In the lamina propria, dendritic cells and macrophages, after their interaction with intestinal microbes, determine the activation of innate immune cells through the release of numerous cytokines (neutrophils, eosinophils, etc.) and trigger the adaptative (Th1 and Th17) immune cells differentiation in mesenteric lymph nodes. Activated T cells, through a vascular homing mediated by the alfa4-beta7–MAdCAM-1 pathway, reach the intestinal lamina propria and spread the inflammatory process. The persistence of inflammation can lead to carcinogenesis and metastasis. Proteins involved in maintaining gut barrier function, such as intestinal fatty acid binding protein and tight junction proteins (shown in the dotted-line circle box in the upper left corner of the figure), have been proposed as potential biomarkers for cancer detection. Created with “Biorender.com”. Abbreviations: E. coli, Escherichia coli; ETBF, Enterotoxigenic Bacteroides fragilis; iFABP, intestinal fatty acid binding protein; IFN, interferon; IL, interleukin; JAM, junctional adhesion molecule; MAdCAM, mucosal vascular addressin cell adhesion molecule; pks+, polyketide synthase productor; SCFA, small chain fatty acid; TNF, tumour necrosis factor.
Under physiological conditions, if epithelial barrier function is preserved, commensal bacteria interact with pattern recognition receptors (PRRs) in the apical membrane of enterocytes, activating a process of microbial tolerance through the inhibition of NF-kB signalling and the induction of a tolerogenic phenotype of dendritic cells (DCs) and macrophages [25][26]. Conversely, in the case of dysbiosis and intestinal barrier impairment, the activation of PRRs on basolateral membrane determines the activation of the inflammatory pathway [27][28]. More in-depth, the interaction of microbial antigens with lamina propria DCs and macrophages directs the inflammation through the production of inflammatory cytokines (TNF-α, IL-12 and IL-23) and the consequent activation of innate immune cells, such as neutrophils and eosinophils. In addition, the interaction between DCs and microbes activates the adaptative (Th1 and Th17) immune response in mesenteric lymph nodes. Through the vascular system, activated T cells reach the lamina propria, spreading inflammation by producing additional inflammatory cytokines (such as IFN-gamma, IL-17, and IL-22). As previously discussed, the persistence of inflammation can lead to carcinogenesis and metastasis.
5.1. Microbiome Interaction in Colorectal Cancer Development
In recent years, the role of bacteria in the carcinogenic process has been extensively explored in several studies through in vitro cell culture, intestinal organoid models, and mouse models of inflammation.
Immuno-deficient mouse models of CRC supported the mechanisms of microbiota-induced inflammatory tumorigenesis and susceptibility between individuals depending on Toll-Like Receptor (TLR)/MyD88 and inflammasome signalling [29].
Currently, three bacterial species have been linked in particular to the process of human colorectal carcinogenesis: Fusobacterium nucleatum (Fn), Escherichia coli containing pathogen polyketide synthetase (pks) islands, and Bacteroides fragilis expressing B. fragilis toxin (BFT) [6].
The intestinal microbiota of patients with IBD demonstrates a greater abundance of Enterobacteriaceae/E. coli and patients with IBD and CRC have an increased prevalence of mucosa-associated E. coli compared with non-IBD and non-CRC patients.
E. coli strains that contain the pks gene cluster have been found more often in biopsies from CRC (67%) and IBD (40%) than in healthy control subjects (21%). These results in oncogenic phenotypes manifest in WNT independence and increased proliferation. The pks encodes colibactin, a genotoxin that can stimulate tumour growth. In mucosal inflammation, pks+ E. coli promoted DNA damage and neoplastic transformation in a mouse model [30]. This suggests a direct link between colibactin exposure and increased cancer risk.
Moreover, Enterotoxigenic Bacteroides fragilis expresses the pathogenic BFT which binds to a specific colonic epithelial cell receptor, thus activating Wnt and NF-kB signalling pathways that determined increased cell proliferation, the epithelial release of pro-inflammatory mediators, and DNA damage. In 90% of patients with sporadic CRC, the BFT gene sequences were observed compared with 55% of controls and in the stool of approximately 14% of patients with IBD. Of note, BFT induces acute and chronic colitis in mice and it promotes IL-17-dependent colon carcinogenesis in the Min Apc+/− mouse model [31][32].
Furthermore, metabolites such as short-chain fatty acids produced by the commensal GI microbiota modulate immune cell activation, inflammatory responses, and carcinogenesis via tumour suppressor gene expression and regulatory T-cell proliferation through histone deacetylase inhibition [16]. Other metabolic classes and pathways significantly dysregulated in CRC include bile acids [33]. Experimental mouse models have shown that elevated secondary bile acid concentrations promote intestinal tumorigenesis [16]. Wirbel et al. [34] found the bile acid-inducible (bai) operon to be highly abundant in the stool of CRC patients. They confirmed this finding at both the genomic and transcriptomic levels using qPCR. Moreover, they supported a link between high dietary fat intake and CRC [35]. Together, all these data strongly support the promising role of microbiome-based CRC diagnostics. In the future, identifying disease-specific microbiome signatures for both IBD and CRC, together with metagenomics sequencing and other culture-independent technologies, may be helpful for personalised, early, and noninvasive CRC screening in IBD patients [36].
5.2. Markers of Gut Barrier Functionality for the Early Detection of CRC
Considering the main role of intestinal barrier disruption in CRC development, markers evaluating its function have been considered for early cancer detection.
Proteins involved in maintaining gut barrier function, such as intestinal fatty acid binding protein iFABP, have been proposed as potential biomarkers for detecting early-stage colorectal cancer (CRC) or assessing the malignant potential of adenomas [37]. iFABP is found in intestinal epithelial cells and its leakage into circulation during intestinal damage can upregulate its expression. Interestingly, plasma levels of iFABP are higher in patients with severe UC than in those with mild disease [38].
In addition to iFABP, tight junction proteins such as claudins (CLDNs) and junctional adhesion proteins (JAMs) have also been implicated in the pathogenesis of gastrointestinal cancers [39]. The overexpression of CLDNs has been linked to neoplastic transformation in premalignant epithelial tissue, while lower expression of junctional adhesion proteins (JAM2) is associated with CRC progression, metastasis, and poor prognosis [40]. Examining and identifying colonic mucosal barrier integrity markers may aid the early detection and prediction of the progression of CRC-associated IBD. Probe confocal laser endomicroscopy (pCLE) is an in vivo histology imaging tool capable of assessing ultrastructural and dynamic changes in the intestinal barrier consisting of epithelial cells connected by tight and adherent junctions.
This technique could predict the therapeutic response, thus paving the way to precision medicine [41]. The combination of innovative endoscopic techniques able to study intestinal anatomy and cell function as well as new OMICs techniques, termed “Endo-Omics, will in the future enable a better understanding of IBD, the optimisation of therapeutic management, and cancer prevention [42].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15082389
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434.
- Zhao, M.; Gönczi, L.; Lakatos, P.L.; Burisch, J. The Burden of Inflammatory Bowel Disease in Europe in 2020. J. Crohn’s Colitis 2021, 15, 1573–1587.
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535.
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104.
- Bye, W.A.; Ma, C.; Nguyen, T.M.; Parker, C.E.; Jairath, V.; East, J.E. Strategies for Detecting Colorectal Cancer in Patients with Inflammatory Bowel Disease: A Cochrane Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2018, 113, 1801–1809.
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730.e3.
- Itzkowitz, S.H.; Yio, X. Inflammation and Cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Liver Physiol. 2004, 287, G7–G17.
- Frick, A.; Khare, V.; Paul, G.; Lang, M.; Ferk, F.; Knasmüller, S.; Beer, A.; Oberhuber, G.; Gasche, C. Overt Increase of Oxidative Stress and DNA Damage in Murine and Human Colitis and Colitis-Associated Neoplasia. Mol. Cancer Res. 2018, 16, 634–642.
- Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 2021, 21, 1325.
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144.
- Rajamäki, K.; Taira, A.; Katainen, R.; Välimäki, N.; Kuosmanen, A.; Plaketti, R.-M.; Seppälä, T.T.; Ahtiainen, M.; Wirta, E.-V.; Vartiainen, E.; et al. Genetic and Epigenetic Characteristics of Inflammatory Bowel Disease–Associated Colorectal Cancer. Gastroenterology 2021, 161, 592–607.
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e6.
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216.
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356.
- Sudhakar, P.; Alsoud, D.; Wellens, J.; Verstockt, S.; Arnauts, K.; Verstockt, B.; Vermeire, S. Tailoring Multi-omics to Inflammatory Bowel Diseases: All for One and One for All. J. Crohn’s Colitis 2022, 16, 1306–1320.
- Pratt, M.; Forbes, J.D.; Knox, N.C.; Bernstein, C.N.; Van Domselaar, G. Microbiome-Mediated Immune Signaling in Inflammatory Bowel Disease and Colorectal Cancer: Support From Meta-omics Data. Front. Cell Dev. Biol. 2021, 9, 3288.
- The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease. Cell Host Microbe 2014, 16, 276–289.
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662.
- The Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648.
- Lazaridis; Konstantinos, N. Dissecting the Pathogenesis and Outcomes of PSC Using Multi-Omics by Studying the Exposome and Genome. Available online: https://grantome.com/grant/NIH/RC2-DK118619-03 (accessed on 2 April 2023).
- Zheng, H.-H.; Jiang, X.-L. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2016, 28, 383–390.
- de Krijger, M.; Carvalho, B.; Rausch, C.; Bolijn, A.S.; Delis-van Diemen, P.M.; Tijssen, M.; van Engeland, M.; Mostafavi, N.; Bogie, R.M.M.; Dekker, E.; et al. Genetic Profiling of Colorectal Carcinomas of Patients with Primary Sclerosing Cholangitis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 1309–1320.
- Amoroso, C.; Perillo, F.; Strati, F.; Fantini, M.; Caprioli, F.; Facciotti, F. The Role of Gut Microbiota Biomodulators on Mucosal Immunity and Intestinal Inflammation. Cells 2020, 9, 1234.
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584.
- Sansonetti, P.J. War and peace at mucosal surfaces. Nat. Rev. Immunol. 2004, 4, 953–964.
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153.
- Yu, S.; Sun, Y.; Shao, X.; Zhou, Y.; Yu, Y.; Kuai, X.; Zhou, C. Leaky Gut in IBD: Intestinal Barrier-Gut Microbiota Interaction. J. Microbiol. Biotechnol. 2022, 32, 825–834.
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809.
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867.
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 4724.
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis Toxin Gene Is Prevalent in the Colon Mucosa of Colorectal Cancer Patients. Clin. Infect. Dis. 2015, 60, 208–215.
- Prindiville, T. Bacteroides fragilis Enterotoxin Gene Sequences in Patients with Inflammatory Bowel Disease. Emerg. Infect. Dis. 2000, 6, 171–174.
- Peng, Y.; Nie, Y.; Yu, J.; Wong, C.C. Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications. Metabolites 2021, 11, 159.
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689.
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164.
- Pratt, M.; Forbes, J.D.; Knox, N.C.; Van Domselaar, G.; Bernstein, C.N. Colorectal Cancer Screening in Inflammatory Bowel Diseases—Can Characterization of GI Microbiome Signatures Enhance Neoplasia Detection? Gastroenterology 2022, 162, 1409–1423.e1.
- Genua, F.; Raghunathan, V.; Jenab, M.; Gallagher, W.M.; Hughes, D.J. The Role of Gut Barrier Dysfunction and Microbiome Dysbiosis in Colorectal Cancer Development. Front. Oncol. 2021, 11, 626349.
- Bottasso Arias, N.M.; García, M.; Bondar, C.; Guzman, L.; Redondo, A.; Chopita, N.; Córsico, B.; Chirdo, F.G. Expression Pattern of Fatty Acid Binding Proteins in Celiac Disease Enteropathy. Mediat. Inflamm. 2015, 2015, 738563.
- Kinugasa, T.; Akagi, Y.; Yoshida, T.; Ryu, Y.; Shiratuchi, I.; Ishibashi, N.; Shirouzu, K. Increased claudin-1 protein expression contributes to tumorigenesis in ulcerative colitis-associated colorectal cancer. Anticancer Res. 2010, 30, 3181–3186.
- ZHAO, H.; YU, H.; MARTIN, T.A.; ZHANG, Y.; CHEN, G.; JIANG, W.G. Effect of junctional adhesion molecule-2 expression on cell growth, invasion and migration in human colorectal cancer. Int. J. Oncol. 2016, 48, 929–936.
- Nardone, O.M.; Cannatelli, R.; Ghosh, S.; Iacucci, M. New endoscopic tools in inflammatory bowel disease. United Eur. Gastroenterol. J. 2022, 10, 1103–1112.
- Iacucci, M.; Jeffery, L.; Acharjee, A.; Grisan, E.; Buda, A.; Nardone, O.M.; Smith, S.C.L.; Labarile, N.; Zardo, D.; Ungar, B.; et al. Computer-Aided Imaging Analysis of Probe-Based Confocal Laser Endomicroscopy With Molecular Labeling and Gene Expression Identifies Markers of Response to Biological Therapy in IBD Patients: The Endo-Omics Study. Inflamm. Bowel Dis. 2022, 2022, 1–12.
This entry is offline, you can click here to edit this entry!