Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Renal cancer is a group of several tumors that develop in the kidney, each with a unique histology and clinical evolution, each responding differently to treatment, and each determined by a different gene mutation. A wide range of cellular processes, including survival, proliferation, growth, metabolism, angiogenesis, and metastasis, are regulated by the phosphatidylinositol-3-kinase PI3K/AKT/mammalian target of rapamycin (mTOR) signaling pathway, which is overactivated in different cancer types by molecular abnormalities.
- PI3K/AKT/mTOR
- renal cancer
- metabolism
1. Renal Cancer Types and Renal Cancer Incidence
Renal cancer is a group of several tumors that develop in the kidney, each with a unique histology and clinical evolution, each responding differently to treatment, and each determined by a different gene mutation. Recently, the crucial role that metabolic pathways play in cancer has been emphasized. Moreover, research shows that the genes whose mutations are associated with renal cancer have interactions with the cell metabolism pathways for energy, nutrition, iron, or oxygen sensing [1].
Classically, any tumor is described on a histological basis. The World Health Organization postulates the shift of the diagnostic/classification criteria of renal tumors from exclusive morphologic criteria towards combined (integrated) criteria: clinical, imagistic, histologic, immunohistochemistry, and molecular (molecular markers, proteomics, study of the tumor microenvironment) [2][3][4].
Morphological criteria, which include the anatomical location (collecting duct carcinomas, juxtaglomerular cell tumors, renal medullary carcinomas) and the histology by cytoplasmic coloration (clear cell renal carcinomas, eosinophilic renal carcinomas, chromophobe renal carcinomas), cell form (spindle cell carcinoma), histological type of tissue involved in the tumoral process (epithelial renal cancers, stromal renal cancers, mesenchymal renal cancers) can be used to define and classify renal tumors [5].
Molecular and genetic criteria such as loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinomas [6][7], genetic abnormalities in renal cell carcinomas, involving chromosomes 7 and 17 [8][9][10][11], multiple chromosomal losses and/or abnormalities in chromophobe renal cell carcinomas [12][13] are also included to define and classify renal malignancies.
Additionally, renal cancer can be described based on the existence of correlations with specific renal diseases (acquired cystic disease–associated renal carcinomas) or with syndromes that have familial aggregation (hereditary leiomyomatosis and renal cell carcinoma syndrome–associated renal cell carcinoma) [14][15]. Table 1 presents the latest combined IACR/IARC/WHO classification of renal tumors [5].
Table 1. IACR/IARC/WHO classification of renal tumors.
| Renal Cell Tumors | |
|---|---|
| Clear Cell Renal Tumors | |
| 8310/3 | Clear cell renal cell carcinoma |
| 8316/1 | Multilocular cystic renal neoplasm of low malignant potential |
| Papillary renal tumors | |
| 8260/0 | Papillary adenoma |
| 8260/3 | Papillary renal cell carcinoma a |
| Oncocytic and chromophobe renal tumors | |
| 8290/0 | Oncocytoma |
| 8317/3 | Chromophobe cell renal carcinoma |
| Other oncocytic tumors of the kidney | |
| Collecting duct tumors | |
| 8319/3 | Collecting duct carcinoma |
| Other renal tumors | |
| 8323/1 | Clear cell papillary renal cell tumors |
| 8480/3 | Mucinous tubular and spindle cell carcinoma |
| 8316/3 | Tubulocystic renal cell carcinoma |
| 8316/3 | Acquired cystic disease–associated renal cell carcinoma |
| 8311/3 | Eosinophilic solid and cystic renal cell carcinoma |
| 8312/3 | Renal cell carcinoma, NOS |
| Molecularly defined renal carcinomas | |
| 8311/3 | TFE3-rearranged renal cell carcinomas |
| 8311/3 | TFEB-altered renal cell carcinomas |
| 8311/3 | ELOC (formerly TCEB1)-mutated renal cell carcinoma |
| 8311/3 | Fumarate hydratase–deficient renal cell carcinoma |
| 8311/3 | Hereditary leiomyomatosis and renal cell carcinoma syndrome–associated renal cell carcinoma |
| 8311/3 | Succinate dehydrogenase–deficient renal cell carcinoma |
| 8311/3 | ALK-rearranged renal cell carcinomas |
| 8510/3 | Medullary carcinoma, NOS |
| 8510/3 | SMARCB1-deficient medullary-like renal cell carcinoma |
| 8510/3 | SMARCB1-deficient undifferentiated renal cell carcinoma, NOS |
| 8510/3 | SMARCB1-deficient dedifferentiated renal cell carcinomas of other specific subtypes |
| Metanephric tumors | |
| 8325/0 | Metanephric adenoma |
| 9013/0 | Metanephric adenofibroma |
| 8935/1 | Metanephric stromal tumor |
| Mixed epithelial and stromal renal tumors | |
| 8959/0 | Mixed epithelial and stromal tumor |
| 8959/0 | Adult cystic nephroma |
| 8959/0 | Pediatric cystic nephroma |
| Renal mesenchymal tumors | |
| Adult renal mesenchymal tumors | |
| 8860/0 | Angiomyolipoma |
| 8860/0 | Oncocytic angiomyolipoma |
| 8860/0 | Angiomyolipoma with epithelial cysts |
| 8860/1 | Angiomyolipoma, epithelioid |
| 9161/1 | Hemangioblastoma |
| 8361/0 | Juxtaglomerular tumor |
| 8361/0 | Functioning juxtaglomerular cell tumor |
| 8361/0 | Nonfunctioning juxtaglomerular cell tumor |
| 8966/0 | Renomedullary interstitial cell tumor |
| Pediatric renal mesenchymal tumors | |
| 8967/0 | Ossifying renal tumor of infancy |
| 8960/1 | Mesoblastic nephroma |
| 8960/1 | Classic congenital mesoblastic nephroma |
| 8960/1 | Cellular congenital mesoblastic nephroma |
| 8960/1 | Mixed congenital mesoblastic nephroma |
| 8963/3 | Malignant rhabdoid tumor of the kidney |
| 8964/3 | Clear cell sarcoma of the kidney |
| Embryonal neoplasms of the kidney | |
| Nephroblastic tumors | |
| Nephrogenic rests | |
| Perilobar nephrogenic rests | |
| Intralobar nephrogenic rests | |
| Nephroblastomatosis | |
| 8959/1 | Cystic partially differentiated nephroblastoma |
| 8960/3 | Nephroblastoma |
| Miscellaneous renal tumors | |
| Germ cell tumors of the kidney | |
| 9084/0 | Prepubertal-type teratoma |
| 9084/3 | Teratoma with carcinoid (neuroendocrine tumor) |
| 9071/3 | Yolk sac tumor, NOS |
| 9085/3 | Mixed teratoma–yolk sac tumor |
This combined IACR/IARC/WHO classification of the renal tumors tends to integrate multiple criteria, including the behavior of the tumors [5]: 0—benign tumors; 1—uncertain, unspecified, or borderline behavior; 2—grade III intraepithelial neoplasia and carcinoma in situ; 3—primitive malignant tumors; 6—metastatic malignant tumors.
1.1. Renal Cell Carcinoma (RCC)
RCC develops from the renal tubular epithelium or the renal cortex [16]. It represents 85–95% of kidney cancers, being the most frequent type of renal cancer in adult patients [17]. It accounts for 3% of all cancer cases and is in 7th place among the most frequent histological types of cancer [18] and worldwide affects over 400,000 patients every year [19]. North America and the Czech Republic are reported to have the highest incidence rates [20]. RCC was discovered at any age, but the sixth decade of life seems to be the most affected [20]. It concerns more frequently in men than women (2/1 ratio) [21] and includes more than 10 histological and molecular subtypes of renal cancers [22].
The major types of RCC are considered those with over 5% incidence each: clear cell renal cell carcinoma (ccRCC), papillary renal cell carcinoma (PRCC), and chromophobe renal cell carcinoma (ChRCC) [23][24][25][26]. The rare types of RCC account for less than 1% of incidence each [22]. There are, also, RCCs that do not comply with the histologic diagnostic criteria of the above-mentioned types (major types or rare types); these are called unclassified renal cell carcinomas (uRCCs) and they have all together less than 4% incidence [27][28].
1.1.1. Clear Cell Renal Cell Carcinoma (ccRCC)
ccRCC represents 75% of the RCCs and accounts for most kidney cancer-related deaths. Moreover, one-third of the patients with ccRCC develop metastases [24][29][30]. It involves patients in the 5th–7th decades of life [29][30]. Some of the ccRCCs associate with genetic disorders, the most prevalent being the von Hyppel-Lindau (VHL) gene (located on the short arm of chromosome 3) and the protein polybromo-1 (PBRM-1) gene (also located on the short arm of the chromosome 3). Apparently, 95% of the ccRCCs exhibit a deletion in 3p (the short arm of chromosome 3) [31]. In addition to the genetic factors, there are acquired factors incriminated in the development of ccRCCs, such as smoking, long-term dialysis, obesity, diabetes mellitus, chronic administration of analgesic medication, and arterial hypertension [30].
ccRCCs develop from the proximal nephron and tubular epithelium [30] and appear as yellowish solid tumors with internal bleeding, necrosis, calcifications, and cystic degeneration. Because the cytoplasm contains large amounts of glycogen and lipids, the histological aspect is clear cells [32][33]. The morphological characteristics (histological grade, tumor size, tumor necrosis, tumoral grading, vascular invasion) are currently the most important prognostic factors of survival in ccRCCs [34].
1.1.2. Papillary Renal Cell Carcinoma (PRCC)
PRCC accounts for 15% of all renal cancers, being the second most frequent kidney malignancy after ccRCC [35]. PRCC develops in the nephron, from the tubular epithelium, usually after the fifth decade of life. The prognosis of PRCC is similar to that of ccRCC [36].
Histologically, PRCC has papillae with vascular cores, foamy histiocytes, and psammoma bodies (round microscopical calcifications) [35]. Papillary tumors appear as long finger-like cell proliferations [35]. Classically, based on staining characteristics, PRCCs are divided into Type 1 (with basophilic cytoplasm) and Type 2 (with eosinophilic cytoplasm) [35][37].
Type 1 PRCC is characterized by gains of 7, 8q, 12q, 16p, 17, and 20 chromosomes and loss of 9p chromosome [11]. Frequently, the mutated gene is MET proto-oncogene (the MET tyrosine kinase domain is altered) [11], located on chromosome 7. Basophilic tumors are classified as low-grade [38].
Type 2 PRCC is characterized by gains of 8q chromosome, and loss of 1p and 9p chromosomes [11]. Frequently, the mutated genes are CDKN2A gene (cyclin-dependent kinase inhibitor 2A), SETD2 gene (SET domain-containing 2)-the SET domain is a protein domain with methyltransferase activity, and NRF2 gene (nuclear factor E2-related factor 2) [11]. Eosinophilic tumors are classified as high-grade [38].
Subclassification of PRCC into Type 1 and Type 2 appears to be no longer recommended and tumors previously classified as PRCC type 2 are presently considered individual entities [39].
1.1.3. Chromophobe Renal Cell Carcinoma (ChRCC)
ChRCC represents about 5% of RCCs [40] and develops in the distal regions of the nephron. This type of RCC has a better prognosis than ccRCC [41], with its’ mortality being around 10%. It is diagnosed more frequently after the fifth decade of life [40].
Histologically, there is the classic type ChRCC (tumor cells exhibiting prominent membrane and pale cytoplasm) and the eosinophilic type ChRCC (large tumor cells with cytoplasm containing fine eosinophilic granules) [40]. Both types present similar cytogenetic alterations, such as the loss of chromosomes 1, 2, 6, 10, 13, 17, and 21, the mutated genes are TP53 gene (Tumor Protein P53 gene) and PTEN gene (Phosphatase and TENsin homolog gene) [41].
1.1.4. Clear Cell Papillary Renal Cell Carcinoma (ccpRCC)
ccpRCC is the 4th most prevalent type of RCCs and is diagnosed more frequently after the 7th decade of life. The clinical course of the tumor is considered to be indolent [42].
Histologically, it has specific features such as low-grade nuclei arranged horizontally and apically, distant from the basal membrane, but also morphological aspects similar to ccRCC and other morphological traits similar to PRCC [42]. It is agreed that specific nuclear arrangements associated with papillary architecture (focal or diffuse) are indicative of ccpRCC [43][44].
ccpRCC was renamed clear cell papillary renal cell tumor (ccPRCT) [39]. This switch from carcinoma toward tumor was performed because of the indolent behavior of this group of tumors [39][42]. The absence of recurrent cytogenetic abnormalities or the absence of VHL gene alterations is differentiation criteria from ccRCCs and PRCCs.
1.1.5. Collecting Duct Carcinoma (CDC)
CDC are rare tumors, representing 2% of all renal malignancies [45]. This type of renal cancer develops between the 2nd–9th decades of life, being reported more frequently in men (men:women ratio is 2:1). It develops from the epithelium of the renal collecting ducts, and it is highly aggressive, having a mortality rate of 70% in two years [18]. Moreover, 71% of the patients with kidney-related clinical signs (the most typical being macroscopic hematuria) already have metastases [45].
Histologically, CDC is relatively polymorphic: tubulopapillary pattern/cords or tubular structures/columnar pattern/desmoplastic stroma/mucinous material [18][46]. CDC presents as cytogenetic alterations gains at chromosome 13q and losses of chromosomes 8p, 16p, 1p, and 9p [18][46]. However, generally, CDC is defined by a “lack of consistency and specificity of molecular profile” [46].
1.1.6. Renal Medullary Carcinoma (RMC)
Renal medullary carcinoma is very rare, representing 0.5–1% of the RCCs [30][47] and it develops more frequently in the 2nd–3rd decades of life, with a higher incidence in Mediterranean and African males [48]. Additionally, it has been observed that patients with sickle cell disease are at increased risk to develop RMC [47][48][49]. This type of carcinoma develops from the distal nephron and is extremely aggressive, with the 3-year survival rate being less than 5% [47][50].
Histologically, RMC is very heterogenous, presenting many patterns—glandular, tubular, tubulopapillary, microcystic, adenoid cystic-like, and reticular—as poorly differentiated eosinophilic cells and inflammatory infiltrative cells [18][47][50].
The affected genes in RMC are SMARCB1 (Hsieh, JJ 2017) and INI1 [47]. The SMARCB1 gene codifies the SMARCCB1 protein (related matrix-associated actin-dependent regulator of chromatin subfamily B member 1), which is a core subunit of the SWI/sucrose non-fermenting (SNF) ATP-dependent chromatin remodeling complex, that relieves repressive chromatin structures [18].
On the other hand, the INI1 gene also loses its expression. INI1 codifies the INI1 protein (Integrase Interactor 1), a protein capable of interacting with the integrase protein of the human immunodeficiency virus. SMARCB1/INI1 is one of the core subunit proteins of the ATP-dependent SWI/SNF chromatin remodeling complex [47] and it has an important tumor suppressor action [51][52].
1.1.7. Unclassified Renal Cancers (uRCCs)
uRCCs represent 3–5% of the RCCs [30] and have a high mortality rate [18], affecting any age [30]. Histologically, uRCCs might share characteristics with other RCCs, or do not resemble any other type [53]. The genomic alterations encountered in uRCCs might be identified in other RCCs, but there are also rare subtypes with particular altered genes [54].
1.2. Urothelial Carcinoma (UC)
Urothelial carcinoma is a cancer that develops in the urothelium and accounts for almost 90% of all cases of bladder cancer and 7% of all cases of kidney cancer, including cancer of the renal pelvis and the ureter [55]. Urinary bladder urothelial carcinoma (UBUC) presents micropapillary sarcomatoid, squamous, and glandular variations. It is accessible to TUR (transurethral resection) and local chemotherapy; hence, it has a good prognosis [56][57][58].
1.3. Nephroblastoma or Wilms’ Tumor (WT)
Nephroblastoma is the most frequent kidney cancer in children and the most frequent abdominal cancer in children [59][60]. It is extremely rarely encountered in adults, the highest incidence being in the first decade of life (between 2 and 5 years of age) [61]. The highest incidence is in Africans and African Americans, while the lowest incidence is in East Asians (who also have a better prognosis). The incidence in North America and Europe is almost the same [61].
Wilms’ tumor may develop due to genetic alterations. Sometimes, the tumor is associated with specific groups of other signs and symptoms, resulting in a syndrome [30][60].
Frequently, the affected genes are WT1 and WT2. WT1 gene (Wilms’ Tumor 1 gene) is located on chromosome 11p13 and is essential for normal embryologic development of the kidney and genitourinary tract. This gene is deleted in WAGR syndrome (Wilms’ tumor, aniridia, genitourinary anomalies, intellectual disability) [62][63]. WT1 gene is muted in Denys–Drash syndrome (Wilms’ tumor, gonadal dysgenesis, nephropathy) [62][63]. At least 50% of the Wilms’ tumors with affected WT1 gene display acquired somatic mutations in CTNNB1 gene (Catenin Beta 1 gene), which is located on chromosome 3 (3p22.1) [64].
WT2 gene (Wilms’ Tumor 2 gene) is located on chromosome 11p15. The abnormal regulation of chromosome 11p15.5 is related to Beckwith–Widemann syndrome (macrosomia, macroglossia, hemihypertrophy, omphalocele, visceromegaly, Wilms’ tumor) [65][66]. It must be noted that most cases of Wilms’ tumor do not exhibit mutations of the above-mentioned genes [67].
Wilms’ tumor develops, in most situations, from metanephrogenic tissue, with metanephrogenic cells being found in 35% of unilateral and 100% of bilateral Wilms’ tumors [68]. The histological type is the most important prognostic factor in Wilms’ tumor [69]. Based on the cell types, there are two histological types of nephroblastoma, the classical type, with a favorable prognosis, and the anaplastic type, with an unfavorable prognosis.
The classical type nephroblastoma accounts for 92–95% of Wilms’ tumors and, due to its favorable histology, has a good prognosis (with correct treatment) [69]. It has a triphasic histological pattern, including metanephrogenic blastema derivatives, epithelial derivatives, and stromal derivatives, in variate proportions [70][71]. Metanephrogenic blastema represents the less differentiated and most malignant component.
The anaplastic type nephroblastoma accounts for 5–8% of Wilms’ tumors and, due to its unfavorable histology, has a poor prognosis [70][71]. The anaplasia is defined by nuclear enlargement, multiple mitoses, pleomorphism, and hyperchromatic nuclei [72][73]. The anaplasia can be focal (a localized area with anaplastic features, surrounded by non-anaplastic cells) or diffuse (all the other situations) [72][73].
In high-income countries, with adequate treatment, the 5-year survival rate in children with a classical Wilms’ tumor is 92% (Northern America). At the same time, in low-income countries, the 5-year survival rate drops to 78% [61]. The anaplastic Wilms’ tumor has a worse prognosis because of its increased chemoresistance [74][75].
1.4. Renal Sarcoma
Renal sarcoma is a rare kidney tumor, accounting for 1–2% of renal malignant tumors [76]. It develops (initially) in the connective tissue and renal blood vessels (renal capsule and perisinuous space). The origin of renal sarcomas is represented by the mesenchymal cells, which is why they expand easily and cross anatomical boundaries (capsulae, fasciae) [76].
Histological types are represented by leiomyosarcomas (LMSs) (50–60% of renal sarcomas), liposarcomas (10–15% of renal sarcomas), and rare variants such as Ewin’s sarcoma/primitive neuroectodermal tumor (PNET), interdigitating dendritic cell sarcoma (IDCS), malignant hemangiopericytoma, malignant fibrous histiocytoma, angiosarcoma, anaplastic sarcoma, myeloid sarcoma, osteogenic sarcoma, synovial sarcoma, rhabdomyosarcoma (RMS), fibrosarcoma, carcinosarcoma [77].
LMSs develop usually in the 4th–6th decades of life and affect women more frequently. The tumor is unilateral in the majority of reported cases, involving especially the right kidney [78]. The histological aspect of renal sarcomas might resemble other renal cancers, but, classically, there are trabeculae or nests made of plump ovoidal cells, separated by fibrovascular septa [77][78].
Genetically, renal sarcomas are considered to have “chaotic” karyotypes, presenting several genetic alterations. Smaller tumors are associated with the overexpression of PRUNE 2 gene (prune homolog 2 with BCH domain) [79] and thus of having a better prognosis, while in large tumors PRUNE 2 gene is downregulated, which is correlated with a worse prognosis. Alterations of FH (Fumarate Hydrase) gene [80][81] can also be present in renal sarcomas. At the same time, 17q duplication is associated with low-risk metastases, longer survival, and better prognosis [82]. Longer survival was also found to be correlated with 1p33-p32.3 duplications [83]. On the other hand, 1q21.3 duplications are associated with shorter survival [83], while 4q31 and 18q22 deletions are associated with an increased risk of metastases [84]. LMSs (pelvic and retroperitoneal, including kidneys) can display mutations of the MED 12 gene (mediator of RNA polymerase II transcription subunit 12 homolog gene) [85] or overexpression of p16 and p53 tumor suppressor proteins [86].
Overall, the prognosis of renal sarcomas is not easily predictable, because of multiple histological subtypes with variable genetic alterations. Moreover, epidemiological studies suggest that these types of tumors have high metastatic potential and poor prognosis [87].
2. PI3K/AKT/mTOR
2.1. Description of PI3K/AKT/mTOR Signaling Pathway
A wide range of cellular processes, including survival, proliferation, growth, metabolism, angiogenesis, and metastasis, are regulated by the phosphatidylinositol-3-kinase PI3K/AKT/mammalian target of rapamycin (mTOR) signaling pathway, which is overactivated in different cancer types by molecular abnormalities [88][89][90].
The basic biological activity of the PI3K family of lipid kinases is to phosphorylate the 3-hydroxyl group of phosphoinositides [91]. There are three classes (I-III) of PI3K, each with a distinct structure and preferred substrate [91][92]. Class I PI3Ks are heterodimers made up of a regulatory and a catalytic subunit and are represented by p85 and p110 (α, β, and δ) which are included in the class IA group and p101 and p110 γ in the class IB PI3K [93]. These four members of the class I PI3K are encoded by PIK3CA, PIK3CB, PIK3CG, and PIK3CD and facilitate the phosphorylation of PtdIns-4,5-P2 to produce PtdIns-3,4,5-P3 (PI(3,4,5)P3)), which will promote the recruitment of cytoplasmic proteins, acting as a second messenger [94].
Due to its frequent alterations, PIK3CA is most involved in human malignancies related to breast and colon cancer. PIK3CB gene mutation is rarer, although it is expressed ubiquitously. PIK3CD is mostly expressed in white blood cells and B cells and is crucial for the survival and maturation of B cell follicles, while PIK3CG expression is inversely correlated with colon cancer growth, despite the fact that PIK3CG levels are connected to cancer growth [95].
p110α, encoded by PIK3CA on chromosome 3 at 3q26.3, p110β by PIK3CB at 3q23, and p110δ are PI3-kinases that interact with a family of Src homology 2(SH2)-domain-containing regulatory adaptor proteins which facilitate their activation by growth factor receptors [96]. Studies show that p110α isoform can influence the size of the adult heart and has an important role in the survival of the cell, while p110β promotes proliferation [97].
PIK3CG at 7q21.11 encodes p110γ, a catalytic subunit that lacks a p85 binding site, which is the only PI3K class IB member [98] together with its adaptor protein, p101, encoded by PI3K regulatory subunit gene at 17p13.1 [99]. Gβγ subunits and also, possibly, Gα subunits promote activation of class IB, which plays a key role as a modulator of inflammation, in heart contractility and allergies [95][100].
Growth factor stimulation causes PtdIns-3,4,5-P3 to be rapidly produced in healthy cells. Lipid phosphatases, such as the tumor suppressor PTEN, quickly break down this compound and stop PI3K signaling by removing the 3′-phosphate [101][102]. Due to the enhanced activity of oncogenic signaling proteins located upstream of PI3K or due to mutational activation of PI3K itself, cancer cells commonly have elevated levels of PtdIns-3,4,5-P3. Many malignancies also show loss of PTEN function, which increases basal and stimulated PtdIns-3,4,5-P3 abundance by slowing down the second messenger’s turnover rate. Furthermore, PIK3CA and PTEN were discovered to be the second and third most altered genes in human tumors [101].
Upon receptor activation, PI(3,4,5)P3 and PI(3,4)P2 bind to a variety of effectors that have pleckstrin homology (PH) domains and collectively control an intricate signaling network, which can regulate the activity of tiny guanosine triphosphatases and protein kinases that govern cellular adhesion, movement, contraction, and secretion [103].
The core of class II PI3Ks (PI3K-C2α, PI3K-C2 β, and PI3K-C2γ) consists of a central C2 domain, a helical domain, and a bilobal kinase, just like class I and class III PI3Ks. These enzymes can generate both phosphatidylinositol 3-phosphate (PtdIns(3)P) and PtdIns(3,4)P2). One of the three class II PI3K isoforms, class II alpha PI3K (PI3K-C2α), encoded by PI3KC2A at 11p15.5-p14 [96], is expressed in mammals and is crucial for embryonic development. Moreover, PI3K-C2α is essential for clathrin-mediated endocytosis, insulin production and signaling, angiogenesis, and primary cilium function. At the same time, research using animal models linked to pathologies such as cancer, diabetes, vascular disease, and thrombosis with loss of function of this isoform of PI3K class II [104].
Recent studies using super-resolution imaging demonstrated that only PI3K-C2β was strongly colocalized to actin filament (F-actin)-associated clathrin-coated pits and vesicles, despite both PI3K-C2β and PI3K-C2α being co-localized to clathrin-coated pits and vesicles. Based on these findings, it appears that the two class II isoforms participate in clathrin-mediated pinocytosis differently [105]. Another study revealed that mitotic progression is regulated by PI3K-C2β, which is encoded by PIK3C2B at 1q32 [96][106]. PI3K-C2β levels that are higher than normal have been found in primary neuroblastoma tumors and cell lines. A group of variations in the promoter and upstream regions of the PIK3C2B gene, which codes for PI3K-C2β, have been linked to an increased risk of prostate cancer. Additionally, the involvement of PIK3-C2β has also been discovered in lung cancer, ovarian and cervical cancer [107][108]. Moreover, a recent study performed on human breast cancer cell lines and human breast cancer tissues revealed elevated levels of PI3K-C2β, showing that there is a correlation between this class II isoform and the tumor’s proliferative capacity. Additionally, PI3K-C2β downregulation prevented breast cancer metastasis development in vivo and inhibits the invasion of breast cancer cells in vitro [107].
Currently, there is very little information regarding the role of PI3KCAγ in malignancy. Nevertheless, there is research suggesting the involvement of this isoform encoded by PIK3C2G at 12p12 in ovarian cancer, pancreatic ductal adenocarcinoma, and leukemia [96][109].
Class III of PI3Ks has a sole representative, Vps34, an enzyme encoded by PI3KC3 chromosome 18 at position 18q12.3. This PtdIns3-kinase is primarily found on intracellular membranes and was first discovered as a mutation of vesicle-mediated vacuolar protein sorting in yeast. PI3K class III promotes endosome fusion during intracellular trafficking events and is engaged in a variety of intracellular trafficking processes, such as autophagy and phagosome formation, retrograde endosome to Golgi transport, and transport at the nuclear membrane [110][111][112][113]. By attaching to a protein complex made up of a regulatory subunit and a catalytic subunit, PIK3C3 regulates autophagy and macrophage phagocytosis (Table 2) [114].
Table 2. Summary of the roles of PI3K classes.
| PI3K Class | Involved in/Effects | References |
|---|---|---|
| Class I | Second messenger for recruitment of cytoplasmic proteins | [94] |
| Survival and maturation of B cell follicles | [95] | |
| Promote inflammation | [100] | |
| Modulator of heart contractility | [100] | |
| Allergies | [100] | |
| Class II | Embryonic development | [104] |
| Insulin production and signaling | [104] | |
| Angiogenesis | [104] | |
| Primary cilium function | [104] | |
| Mitotic progression | [96][106] | |
| Tumor proliferation and invasion | [107] | |
| Class III | Endosome fusion | [114] |
| Autophagy and phagosome formation | [114] | |
| Macrophage phagocytosis | [114] |
The PI3K pathway’s main effector is AKT (a serine/threonine kinase), which has a number of downstream effectors that control important cellular functions [115][116]. AKT, also known as protein kinase B (PKB), exists in mammals in three different isoforms: AKT1, AKT2, and AKT3. While AKT3 is largely expressed in the brain and has relatively limited expression due to its tissue distribution, AKT1 and AKT2 are abundant in numerous tissues, including pancreatic tissue [117].
These isoforms have distinct and important functions in cancer: AKT2 promotes cell migration and invasion, while AKT3 is linked to hormone independence. Furthermore, AKT2 gene is overexpressed in pancreatic cancer, while breast and prostate cancers present AKT3 gene overexpression [118][119].
By participating in several different signaling pathways in the human body, the mammalian Target of Rapamycin (mTOR) controls cell division, autophagy, and apoptosis, being linked to insulin resistance, osteoporosis, cancer, and rheumatoid arthritis. Furthermore, mTOR modulates gene transcription, as well as protein synthesis, thus influencing the differentiation and proliferation of immune cells [120]. Moreover, it also has a significant impact on tumor metabolism [121].
This protein is a 289-kDa serine-threonine kinase consisting of two multiprotein complexes, known as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [122][123][124]. Due to the different structural characteristics of mTORC1 and mTORC2, cells respond differently to a range of upstream signals and they are activated by different downstream effectors, producing distinct cellular responses [125].
mTORC1 is comprised of the catalytic subunit (mTOR), the Raptor (regulatory-associated protein of mTOR), mLST8 or GβL (mammalian lethal with Sec13 protein 8), PRAS40 (proline-rich AKT substrate 40 kDa), and Deptor (DEP-domain-containing mTOR-interacting protein) [126]. By promoting several anabolic processes, such as the biosynthesis of proteins, lipids, and organelles, and by limiting autophagy, mTORC1 positively regulates cell growth and proliferation.
mTORC2 consists of mTOR, Rictor (mAVO3) (rapamycin-insensitive companion of mTOR), mSIN1 (mammalian stress-activated protein kinase interacting protein), and mLST8 [126][127][128]. Additionally, several less-conserved proteins, including PRR5/Protor, PRR5L, and DEPTOR (the inhibitory protein DEP domain-containing mTOR-interacting protein) have been discovered to interact with mTORC2 [129]. Biological processes such as cell survival, metabolism, proliferation, and cytoskeleton organization involve mTORC2 [126].
2.2. Activation of PI3K/AKT/mTOR in Renal Cancer
One of the most common tumor-related signaling pathways, the PI3K/AKT/mTOR signaling pathway, exhibits aberrant hyperactivation in a range of tumor forms, including breast cancer, colorectal cancer, and RCC, making it a key target for cancer therapy [130].
The molecular oncogenic hallmark of ccRCC is the inactivation of the von Hippel-Lindau tumor suppressor gene, which is followed by the subsequent activation of the hypoxia-inducible factors (HIFs) [131]. Up to 95% of sporadic ccRCC have been shown to have biallelic somatic inactivation of VHL. The VHL gene produces the VHL protein (pVHL), which is a part of the complex involved in the ubiquitination and degradation of HIFs [132], and transcription factors with various roles in the cells are fundamental for the adaptation of cells to oxygen deficiency [133]. The inactivation of VHL is carried out through the loss of the 3p chromosome, promoter methylation or mutations [132].
Deregulation of cyclin D1, a cyclin-dependent kinase cofactor necessary for cell cycle progression, is another effect of VHL function loss in RCC [134]. mTORC1 regulates the translation of cyclin D by phosphorylating two downstream effectors, p70S6kinase (S6K) and the binding protein for eukaryotic initiation factor 4E (4E-BP1) [135]. Furthermore, mTORC1 and mTORC2 regulate the translation of HIF-1α and HIF-2α subunits, respectively. Moreover, research shows that in ccRCCs with substandard VHL activity, both HIF-1 α and HIF-2α subunits are overexpressed, leading to MAPK (mitogen-activated protein kinase) signaling pathway and AKT/mTOR activation [136].
However, in the different types of RCC, other genetic and/or epigenetic events occur, one of them being the overexpression of VEGF (vascular endothelial growth factor) [18]. MAPK (mitogen-activated protein kinase) and PI3K/AKT can be activated directly by VEGF, which interacts with VEGF-R1 and VEGF-R2, tyrosine receptors found in endothelial cells [137].
Apart from VEGF, other growth factors, such as IGF (insulin-like growth factor), PDGF (platelet-derived growth factor), and EGF (epidermal growth factor) bind to the N-terminal extracellular region of their corresponding transmembrane receptor tyrosine kinases (RTKs), causing the RTKs’ cytoplasmic regions and linking molecules to autophosphorylate tyrosine residues [138]. VEGF-R2 and other growth factor receptors, including EGFR and IGFR, in ccRCC cells suffer dimerization and activation due to the overexpression of VEGF, EGF, and IGF. When activated, these tyrosine kinase receptors will activate either the PI3K/AKT/mTOR signaling pathway or the RAS/mitogen-activated protein kinase /extracellular signal-regulated kinase (RAS/MEK/ERK) pathway to increase the production of HIFs, thereby accelerating the growth of the tumor [139]. After its allosteric activation, PI3K is recruited to the RTKs via contacts between the p85 SH2 domains and phospho-Tyr residues on members of the RTK complex [138].
Through interactions of their pleckstrin homology domains with PtdIns(3,4,5)P3 produced by PI3K, AKT and its upstream kinase, PDK1 (3-phosphoinositide-dependent protein kinase-1), are recruited to the inner cell membrane. Subsequently, PDK1 phosphorylates AKT at Thr308. AKT is most effectively activated when mTORC2 and other kinases phosphorylate Ser473 in the regulation hydrophobic region of AKT [140]. Once phosphorylated, AKT is activated and translocates from the cell membrane to different cell compartments where it phosphorylates a variety of downstream substrates. Cell survival, development, metabolism, tumorigenesis, and metastasis are just a few of the many physiologic and pathologic cellular processes that are regulated by activated AKT’s phosphorylation of several substrates [90].
The activation of mTORC1 mediated by AKT promotes protein translation and lipid or nucleotide synthesis. Moreover, AKT phosphorylates and inhibits the GTPase-activating protein for the RHEB (Ras-related small G protein RHEB) and TSC1/TSC2 (tuberous sclerosis complex 1/2) [141].
Moreover, upon activation, activated mTORC1 inhibits apoptosis through its downstream effector proteins such as p70S6K1, allowing the cell cycle to go into the G1 phase [137].
In ccRCC, mutations were also found in several mTOR inhibitors, including PTEN (phosphatase and tensin homolog deleted from chromosome 10) and TSC1/2, highlighting the crucial impact of the mTOR signaling the development of ccRCC. Located on chromosome 10q23.3, tumor suppressor gene PTEN participates in cellular signaling pathways that regulate and control DNA repair, cell growth, proliferation, senescence, and apoptosis, as well as genic mutation [142]. Research related to PTEN, an inhibitor of PI3K is epigenetically reduced in ccRCC, reveals that elevated miR-21 is associated with reduced PTEN expression, favoring cell proliferation and migration [143].
Additionally, a study by Que W et al. showed that reduced PTEN expression is a factor that negatively affects the overall survival of kidney cancer patients. Moreover, a positive correlation was found between the decrease in PTEN expression and the development and severity of renal cancer [142]. On the other hand, Hager M et al. revealed that in the initial stage of renal cell carcinoma, PTEN was underexpressed, but the expression pattern of PTEN was not predictive of the prognosis for patient survival [144].
Mutations in either TSC1 (encodes hamartin) or TSC2 (encodes tuberin) determine loss of the suppressor functions of these genes that are located on chromosomes 9q34 and 16p13, respectively, will activate mTOR, promoting aberrant cellular growth, proliferation, and protein synthesis [145][146]. Apart from activating mTOR, TSC1/TSC2 inactivating mutations also result in increased HIFs translation [147].
Furthermore, current research showed that RCC with leiomyomatous stroma (RCCLS) exhibit frequent TSC1 or TSC2 mutations, and it is thought that ESC (eosinophilic solid cystic)-RCC may be pathognomonic for the mutations of these genes [148].
3. Reprogramming Glucose, Lipid, and Amino Acids Metabolism in Renal Cancer
3.1. Glucose Metabolism in Renal Cancer
RCC is a malignancy of dysregulated metabolism, while mTOR is an important regulator of cell metabolism [1]. It is well known that most patients with cancer have changes in nutrition status [149]. Therefore, the daily consumption of animal fats and red meat is correlated with increased cancer mortality, because these types of food induce the synthesis of IGF1 (insulin growth factor 1), an activator of the mTOR pathway which is associated with tumor progression [150].
Insulin can activate the PI3K signaling pathway, which controls cell metabolism and plays an important role in the proliferation and survival of the cell, ultimately deciding its fate [151]. The insulin-dependent control of systemic and cellular metabolism depends on the PI3K/AKT pathway, with AKT2 representing the primary isoform needed for insulin roles in metabolisms. Serine residues on insulin receptor substrates may be phosphorylated by mTORC1, resulting in their ubiquitylation and proteolytic degradation. Chronically increased levels of insulin may promote the activation of the insulin receptor on preneoplastic cells directly or indirectly by promoting the synthesis of IGF1, with studies performed on xenograft models showing that insulin and IGF1 promote cell proliferation and decrease apoptosis, therefore leading to tumor growth [152].
Moreover, mTORC1 stimulates insulin secretion and fatty acids (FA) oxidation in muscles, while in the liver it stimulates gluconeogenesis and reduces the formation of ketone bodies [153].
The most important isoform of PI3K regarding its role in insulin signaling is PI3Kα, playing an important role in insulin resistance. Meanwhile, inflammation, fatty liver, insulin resistance, and diet-related obesity have been linked to PI3Kγ [154].
Through the nuclear receptor PPAR and signaling via ribosomal protein S6 kinase beta-1 (S6K1 or p70S6 kinase) mTOR regulates adipogenesis, reducing lipolysis and promoting adipocyte clustering, leading to accumulation. mTORC1 regulates the activity of SREBP-1c (sterol regulatory element-binding protein-1c), a transcription factor that stimulates the synthesis of FA and cholesterol. Moreover, mTORC1 stimulates the synthesis of triglycerides by modulating the phosphorylation of a phosphatase lipin 1 (Lpin1) [153].
Metabolic reprogramming plays a pivotal role in cancer progression because it allows the cells to survive in nutrient- and oxygen-deprived conditions, become tolerant to stress, and have the capacity to metastasize in different body sites [155][156].
Moreover, in ccRCC metabolic reprogramming of glucose, fatty acid metabolism and tricarboxylic acid cycle have been identified, with glycolysis being the most frequent metabolic alteration in RCC [157][158]. In renal cancer, reprogramming metabolism takes place in stromal cells [155].
Normally, most mammalian tissues, including the renal cortex with proximal and distal tubules, have an O2 content of 2–9%. Additionally, in normal conditions, our kidneys can metabolize free fatty acids (FFAs), glycerol, lactate, pyruvate, 3-hydroxybutyrate, glutamine, amino acids (proline, glutamine), and Krebs cycle intermediates (citrate, α-ketoglutarate). In the proximal tubule reabsorption of the mentioned compounds occurs in a percentage of 70%. Moreover, FFAs-β-oxidation occurs in the mitochondria-proximal tubule cells [159]. For example, excessive kidney FA loads induce proximal tubular epithelial cells toxicity and lead to kidney disease development such as tissue fibrosis [160].
In contrast, the kidney medulla is physiologically less oxygenated, with 1.4–2.8% of O2 [161]. In kidneys, oxygen is used differently, from the cortex to the medulla. Therefore, a difference in metabolism from the cortical to medullary nephron segments exists. In addition, proximal tubules have a highly aerobic metabolism, where oxidative phosphorylation is used to generate ATP, while the distal nephron has decreased aerobic conditions [162]. In healthy subjects, in kidney tubules, ATP is obtained mostly by fatty acid β-oxidation (FAO) [163].
RCC is characterized by several metabolic dysregulations including oxygen sensing (VHL/HIF pathway), glucose transporters (GLUT1 and GLUT4) energy sensing, and energy nutrient sensing cascade [153]. RCC presents cellular alterations in cellular sugars, lipids, amino acids, and nucleic acids’ metabolism [157][164]. Therefore, dysregulation of cellular metabolism is an important cancer hallmark that contributes to tumor initiation, progression, and tumor heterogeneity [165].
ccRCC is characterized by the loss of the VHL gene and further overexpression of hypoxia-inducible factor 1 alpha (HIF-1α), involved in cancer cell metabolic reprogramming [166]. On the other hand, in the pathogenesis of ccp-RCC, HIF is also activated by non-VHL-dependent mechanisms because immunohistochemical results revealed the presence of HIF-1 and GLUT-1 [167].
Tumors can reprogram metabolic pathways involved in nutrient uptake via a phenomenon called reprogramming. These molecular changes are possible because tumor suppressor genes are lost, and oncogenes are activated. Because multiple metabolic pathways suffer alterations in RCC, this malignant disease is considered a “metabolic disease” [168].
Therefore, ccRCC- metabolic dysregulations involve aerobic glycolysis, Krebs cycle, pentose phosphate pathway, fatty acid synthesis and beta-oxidation, oxidative phosphorylation, glutathione, and glutamine metabolism [169][170][171]. The genes involved in glycolysis, the pentose phosphate pathway, glutamine metabolism, and lipogenesis are upregulated [172]. In cancer cells, the tumor suppressor of HIF, pVHL, will bind and degrade HIF. Moreover, in ccRCC, VHL/pVHL is mutated and its functions are lost, leading to HIF protein overexpression [173]. HIF was also linked to renal medullary carcinoma [174].
In ccRCC, HIF will induce the transcription of genes necessary for glycolysis and lactate metabolism [175]. HIF has two subunits, alpha, and beta. The family of enzymes, prolyl-4-hydroxylases (PHD1–3), catalyze the hydroxylation of proline residues from HIF-alpha subunits, activating hundreds of genes involved in angiogenesis, proliferation, survival, metabolism, apoptosis [176] motility, cytoskeletal structure, cell adhesion, and cellular metabolism [177]. HIFs are O2-sensitive transcription factors that adapt RCC cells to hypoxia, regarding VEGF production, metabolic reprogramming of cellular glucose, and also energy metabolism [178]). In addition, via transcriptional regulation, HIF acts on VEGF, PDFG, EGF, GLUT 1, TGF-alpha, and erythropoietin [179].
Therefore, HIF-1α contributes to cancer cell proliferation, migration, and survival (de Carvalho PA, 2021) and other processes that include GLUT 1 and cyclin D [166]. In normal tubular cells, HIF-1α is present, while HIF-2α is found in pVHL-detective tubular renal cells [136].
HIF-1α protein was detected in VHL-deficient renal epithelial cells, and also ccRCC, suggesting its implications in cancer progression [180]. Therefore, mutations in VHL and alterations in its downstream pathways play crucial roles in RCC development and progression [181]. In addition, hypoxia induces angiogenesis, which plays a significant role in RCC progression [182].
Moreover, HIF activates Ras, leading to Krebs cycle substrate accumulation, including fumarate and also PI3K/AKT/mTOR activation [183]. In RCC, HIF-1α controls cancer cell metabolism, by increasing glucose uptake, through glycolytic and pentose phosphate pathways. In addition, Lucarelli G and his research team detected in tumor tissue increased levels of glucose, and intermediates of upstream glycolytic compounds such as glucose-6-phosphate and fructose-6-phosphate. Instead, the levels of 3-phosphoglycerate, 2-phosphoglycerate, and phosphoenolpyruvate, downstream intermediates were decreased [184].
RAS promotes glycolysis in renal cancer cells because increases the expression of GLUT 1, which further conduces to lactate production by the Warburg effect [185]. Moreover, hyperinsulinemia ensures the energy for tumor cells and sustains their growth [186]. The Warburg effect requires increased glucose levels. For this reason, tumor cells upregulate the expression of glucose transporters (GLUTs) sodium-dependent glucose transporters (SGLTs) [187].
Furthermore, HIFs induce the expression of several proteins and enzymes in glucose metabolism such as GLUT1, LDH, PDK1 (pyruvate dehydrogenase kinase), HK (Hexokinase), and PGK (phosphoglycerate kinase). HIFs also mediate the inhibition of the Krebs cycle and further oxidative phosphorylation [168]. In ccRCC, pVHL invalidation stabilizes HIF in the presence of oxygen, stimulating further glycolytic gene expression [188].
It is considered that VHL inactivation induces in mice and human renal cysts formation but is not enough to cause ccRCC [189].
In most cancer cells and especially in high-grade ccRCC tissues, the Warburg effect is overexpressed [190]. In the presence of oxygen, cancer cells consume glucose and produce lactate. Studies performed in vivo using carbon labeling revealed that transformed cells and T cells use glucose in an anaerobic mode [191]. RCC is correlated with increased production of lactate and nitric oxide (NO) [192]. An elevated level of the enzyme LDHA, implicated in the reduction of pyruvate to lactate was detected in renal cancer cells compared with normal cultures [193].
Glucose-6-phosphate (G6P), an important enzyme used to produce energy as ATP and NADH, is used for glycogenesis and by the pentose phosphate pathway (PPP). The activity of this enzyme is elevated in cells undergoing normal or neoplastic cell growth, including RCC [194]. Mitochondrial metabolism is involved in cancer cell growth, providing precursors to synthesize macromolecules. Therefore, citrate and acetyl-CoA obtained from glucose metabolism are used in tumorigenesis for lipid synthesis [195]. The glycolytic intermediates are used by cancer cells to synthesize lipids, amino acids, and nucleic acids [196]. The isoform pyruvate kinase M2 (PKM2) activity is essential for cancer survival, including RCC, which has been overexpressed in the proximal renal tubule [197].
Cancer cell metabolism is associated with the dysregulation of GLUT1, whose mRNA levels are influenced by microRNAs. In this context, Morais M. and his research team evaluated the levels of intra and extracellular levels of microRNAs and protein levels in three ccRCC levels. The study reported decreased intracellular levels of microRNAs and increased levels of GLUT 1. The study also detected an increased amount of glucose consumption correlated with higher levels of lactate production [198].
AKT plays a key role, in both glycolytic and oxidative metabolism, because its activation will increase ATP production, while its deprivation reduces ATP levels. Moreover, AKT enhances the expression of glucose transporters, increasing the coupling between oxidative phosphorylation and glycolysis, leading to HIF1α and HK2 accumulation, and also activation of phosphofructokinase-2 (PFK-2) [199].
Fumarate hydratase (FH), an enzyme from the Krebs cycle catalyzes the conversion of fumarate to malate. Mutations of this enzyme will lead to malate accumulation, correlated with important metabolic changes [200]. A study by Xin Ge et al. revealed that in patients diagnosed with type 2 papillary renal cell carcinoma, FH deficiency leads to fumarate accumulation, which will inhibit the negative regulator PTEN, therefore promoting tumorigenesis and therapeutic resistance [201]. Moreover, Wang Q and his research team also detected in papillary renal cell carcinoma that the most up-regulated gene was lactate dehydrogenase A (LDHA), while FH is downregulated, both involved in ATP generation [202]. Additionally, hereditary leiomyomatosis and renal cell carcinoma are characterized by germline mutation of the Krebs cycle enzyme, fumarate hydratase [203].
Furthermore, Sudarshan S. and co-workers reported that FH depletion is correlated with acute upregulation of GLUT1 protein, glucose uptake, and further lactic acid production. In addition, succinate accumulation may have similar effects. Taking into consideration these aspects, cancer renal cells will use glycolysis as a source of ATP when TCA enzyme expression is reduced [204]. Moreover, in RCC FH deficiency seems to be correlated with increased levels of HIF-1α and HIF-2α [205].
Increased production of lactic acid is correlated with cancer metastasis, via TGF-β2 pathway. Moreover, lactic acid stimulates endothelial cells to produce VEGF and can generate pyruvate that is converted by decarboxylation into acetyl-CoA, which will be further used by cancer cells for cholesterol and FA synthesis [206].
The pentose phosphate pathway (PPP) is a metabolic pathway where glucose is used not to obtain ATP; instead, it generates ribose 5-phosphate and NADPH. NADPH is necessary for GSH regeneration from GSSG. In ccRCC, a higher GSH/GSSG ratio was detected, important for GSH conversion. Additionally, glucose-6-dehydrogenase, the first enzyme from the pentose pathway is elevated in ccRCC patients [207].
PPP occurs in ccRCC cells to obtain NAPDH and ribose-5-phosphate, used further for nucleotide and lipid biosynthesis. Moreover, NAPDH produced by renal cancer cells is used to fight against reactive oxygen species (ROS) [208].
Increased glycolysis leads to ATP production used for renal cancer cell proliferation. Renal cancer cell-FH deficiency conduces to decreased levels of p53 and increased production of FA biosynthesis because the activity of acetyl-CoA carboxylase is decreased [209].
Furthermore, ccRCC is characterized by increased glycogen and lipid content [210]. It is already known that glycogen represents the mammalian storage of alpha-glucose, bounded 1–4 and 1–6. The liver, kidney, muscle, and brain are the most important energy-consuming organs in normal human physiological processes. In pathological conditions such as cancer, increased glycogen benefits tumor microenvironment development [211]. Schaeffeler E. and co-workers isolated cells from ccRCC and observed that primary cultures are characterized by lipid and glycogen storages and also aerobic glycolysis/lactate fermentation [193].
Glycogen phosphorylase (GP), glycogen synthase (GYS), phosphoglucomutase 1 (PGM1), and protein phosphatase 1 regulatory 3 (PPP1R3) are enzymes involved in glycogen degradation and synthesis. In cancer cells, hypoxia, tumor suppressors, and oncogenes regulate glycogen metabolism, favoring its degradation [212].
Responsible for dysregulation in glycogen metabolism are histone modification [212]. Glycogen synthase kinase-3 (GSK-3) is a key serine/threonine kinase involved in glycogen metabolism that regulates the cell cycle and proliferation. GSK-3 dysregulation leads to its involvement in the pathogenesis of many human diseases such as type 2 diabetes, bipolar disorder, inflammation, and cancer [213]. In hypoxic conditions, oxidative phosphorylation switches to anaerobic metabolism [214].
Taking this into consideration, glycolysis intermediates represent the precursors for lipids, amino acids, and nucleotide synthesis necessary for tumor cell survival and proliferation [215].
3.2. Lipid Metabolism in Renal Cancer
Alteration of the lipid metabolism allows tumor cells to survive [216]. In normal renal proximal tubule cells, FA via beta-oxidation ensures the ATP necessary for renal sodium reabsorption [217]. Fatty acids and cholesterol synthesis were overexpressed in ccRCC and correlated with tumor aggressiveness and poor prognosis [218]. Additionally, lipid droplet formation is a defining histological feature in ccRCC.
Cancer lipid metabolic reprogramming is implicated in the biosynthesis of fatty acids, ceramides, and sphingolipids, which regulates various signaling pathways important for cancer cells. Therefore, cancer cells have increased de novo synthesis and β-oxidation of FFAs [219]. Through HIF-dependent modulation of proteins involved in fatty acids metabolism (uptake, synthesis, usage, storage), in hypoxic conditions, lipogenesis is enhanced. HIF-1 activates the gene that encodes the synthesis of PPARγ (peroxisome proliferator-activated receptor gamma) transcription factor, which stimulates the uptake of FA and the synthesis of TAGs [220]. Furthermore, in cancer cells, HIF-1 directly targets AGPAT2 (acylglycerol-3-phosphate acyltransferase 2), which enhances the TAG biosynthesis pathway, resulting in increased storage of lipids [221].
Młynarczyk G and his research team detected in ccRCC tissues accumulation of sphingosine, sphingosine-1-phosphate (S1P), ceramide, dihydrosphingosine, and dihydroceramide compared with healthy subjects [222]. In ccRCC, lipid accumulation will affect cellular energy homeostasis and lipid signal transduction [219], contributing to cancer cell proliferation, progression, and metastasis [223][224].
In cancer cells, lipolysis is increased leading to an increased amount of FA accumulation and further LDL formation. In renal hypoxic conditions, LDL is accumulated [225].
Li W. and his research team using 10 pairs of cancerous and adjacent normal tissues collected from ccRCC patients detected 28 lipid classes. The study reported that the most abundant lipids with increased levels were triacylglycerol, diacylglycerol, phosphatidylcholine, and phosphatidylethanolamine. Moreover, among them, esterified cholesteryl presented a considerably increased level in tumoral cells compared with normal samples. Activated saturated and unsaturated FA bound by carnitine also had increased levels in tumoral renal cells versus healthy cells [226].
Moreover, in ccRCC increased levels of cholesterol disrupt the lipid metabolism in T cells, leading to the suppression of immune function, and protecting the cells against various treatments [227]. Cholesterol production of cholesterol and synthesis of fatty acids was also found elevated in tumors with WT1 mutations [228].
Dysregulation of the following enzymes—hydroxy acyl-CoA dehydrogenase α-subunit (HADHA), acetyl-CoA acetyltransferase 1 (ACAT1), ATP-citrate lyase (ACLY), and ATP synthase β- subunit (ATP5B)—were also detected in RCC (Liu S/2019). HADHA represents the alpha subunit of the mitochondrial trifunctional protein that catalyzes the last three stages of long chain FFAs-β-oxidation [229].
Additionally, HIFs influence the lipid metabolism of renal cancer cells, acting on the enzyme implicated in the mitochondrial fatty acid transport, carnitine pal-mitoyltransferase 1A (CPT1A), which is the rate-limiting enzyme. Moreover, HIF-1 and HIF-2 repress CPT1A, forcing FA to form droplets for storage [230].
3.3. Amino Acids Metabolism in Renal Cancer
Amino acids are used not only for protein biosynthesis but are also important intermediates for carbon and nitrogen biosynthesis, ATP generation, S-adenosyl methionine, and antioxidant production [231]. Yang H. and colleagues demonstrated that element-binding protein 1 (SREBP1) is overexpressed in ccRCC cell lines, and is important for ccRCC lipid desaturation and cell growth. In addition, SREBP1 activates the NF-kB pathway [232].
The “kidney-type” glutaminase (GLS1) is an enzyme that plays a key role in glutaminolysis [233]. RCC cells utilize glucose and glutamine to support cell growth and proliferation [234].
Glutamine is essential for many cancer cells’ fundamental functions such as the production of antioxidants, maintaining mitochondrial metabolism, and activation of cell signaling. In mitochondria, the dehydrogenases of glutamate to α-ketoglutarate are catalyzed by glutamate dehydrogenase, regulated by ADP-ribosylation, a process mediated by mitochondria protein sirtuin 4 (SIRT4) [235].
Glutamine is one of the most abundant plasma amino acids and an important compound used to produce energy in ccRCC. Glutamine can enter the cell via glutamine transporter (ASCT2) where it can suffer transamination with α-ketoglutarate and NADH formation. Moreover, glutamine can be used for NADPH, ammonium, and other non-essential amino acid production. Further, these compounds derive from glutamine and are used for amino acids, nucleotides, FA, citrate, and oxaloacetate synthesis (Figure 1) [236]. Moreover, glutamine is a donor of nitrogen necessary for nucleotides, hexosamines, and non-essential amino acid biosynthesis [237].
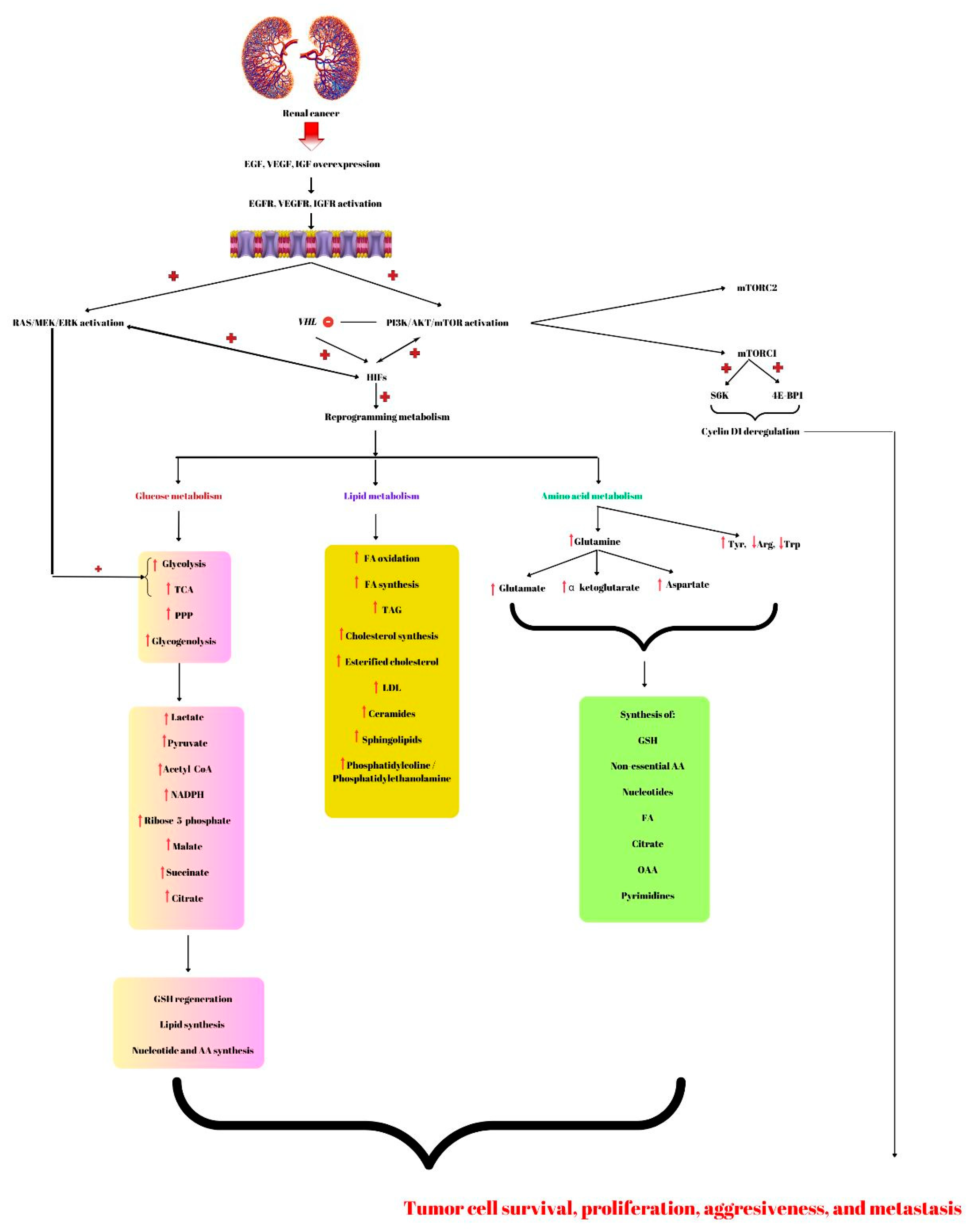
Figure 1. Renal cancer is characterized by overexpression of growth factors; epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), and insulin growth factor (IGF) that will induce the activation of the phosphatidyl-3-kinase-protein-kinase B-mammalian target of rapamycin (PI3K/AKT/mTOR) and RAS/MEK/ERK (RAS/mitogen-activated protein kinase/extracellular signal-regulated kinase). Further, this molecular signaling pathway inhibits the Von Hippel-Lindau (VHL) gene leading to the activation of hypoxia-inducible factors (HIFs) involved in renal cancer-reprogramming metabolism. Therefore, regarding glucose metabolism, this malignancy has increased glycolysis, glycogenolysis, pentose phosphate pathway (PPP), and increased tricarboxylic acid cycle (TCA), leading to lactate, pyruvate, NADPH, ribose-5-phosphate, malate, succinate, citrate, and acetyl-CoA accumulation. Additionally, the RAS/MEK/ERK signaling pathway promotes glycolysis and TCA. The compounds that resulted from altering glucose metabolism are involved in glutathione (GSH) regeneration, lipids, nucleotides, and amino acids (AA) biosynthesis. Fatty acids (FA) are used to produce energy by β-oxidation. Lipid metabolism alterations also include synthesis of FA, TAG (triacylglycerols), cholesterol, ceramides, sphingolipids, and phosphatidylcoline/phosphatidylethanolamine. Amino acids such as glutamine can suffer several modifications forming glutamate, α-ketoglutarate, and aspartate. The intermediates together with increased Tyr (tyrosine) and decreased Arg (arginine) and Trp (tryptophan) will be used by renal cancer cells for the synthesis of new AA, nucleotides, FA, citrate, oxaloacetate (OAA), and pyrimidine bases. All these compounds that result from the altered pathway lead to cancer cell survival, aggressiveness, and metastasis. These characteristics of malignant tumors are also promoted by cyclin D1 deregulation produced by mTORC1 by activating S6K and 4E-BP1. + represents activation; − means inhibited.
From glutamine, aspartate can be obtained, as it is involved in pyrimidine biosynthesis and GSH for cellular redox protection. In addition, glutamine can contribute to cancer resistance against therapy. For example, mTORC1 activation induces glutamine anaplerosis by repressing SIRT4 transcription leading to glutamate dehydrogenase activation [238]. In RCCs, HIFs support and stimulate the reductive glutamine metabolism [239][240]. By inducing the expression of GLS1 (glutaminase 1), HIF-1 augments the level of α-ketoglutarate in cancers, allowing increased citrate synthesis and increased FA/lipid production [241]. Therefore, by acting on AA metabolism, HIFs influence glutamine signaling and enhance tumor progression [242].
Wang J and his research team illustrated that two enzymes involved in tyrosine metabolism, homogentisate 1,2-dioxygenase (HGD), and glutathione S-transferase zeta 1 (GSTZ1) were downregulated in three renal cancer types (KIRC, KIRP, and KICH) of tissues versus healthy ones. In KIRC, these enzymes promote aerobic glycolysis, coordinate amino acid and energy metabolism, and activate the tumor cell cycle, leading to cancer progression [243].
While in healthy status, arginine is a semi-essential or conditionally essential amino acid necessary for development [244][245]. In RCC, amino acids arginine and tryptophan have decreased levels (Yuan Y/2022). Moreover, the mTORC1/4E-BP axis regulates the synthesis of aspartate, asparagine, and serine by modulating mRNA translation [246].
Glutathione, the cell-atypical non-enzymatic tripeptide protects our cells against hydroperoxides, hydrogen peroxide, and lipids peroxides. Miess H. and co-workers reported in the MYC-dependent mouse model of renal cancer that GSH activation blocks tumor growth [247].
The enzyme carbonic anhydrase IX (CA9) is a pH-regulating transmembrane protein, over-expressed in solid tumors, including ccRCC. Xu J and his colleagues confirmed that ccRCC-CA9 mRNA expression was significantly elevated. It was observed that CA9 knockdown upregulates proteins involved in oxidative phosphorylation, and increases mitochondrial biogenesis, leading to the reversal of the Warburg phenotype and in the end cancer cell growth inhibition. The study conducted by Xu J observed that CA9 knockdown upregulated the enzyme mitochondrial arginase 2 (ARG2), resulting in putrescine accumulation. The obtained diamine will further suppress ccRCC proliferation [44].
In WT, numerous other metabolic proteins, such as aldehyde dehydrogenases and proteins involved in propanoate and butanoate metabolism, long-chain fatty acid metabolism, and the breakdown of the branched-chain amino acids valine, leucine, and isoleucine, were also found to be reduced versus normal tissue. It is unclear, though, if these modifications also exist in primary, untreated WT since the tissue samples analyzed by Hammer E et al. were taken after chemotherapy treatment [248].
The metabolic reprogramming in chRCC has been discovered using proteome profiling and includes halted gluconeogenesis, downregulated oxidative phosphorylation, and altered fatty acid and amino acid metabolism. In chRCC, an analogous anticorrelation between transcripts and proteins (as in pRCC) was discovered. Compared to ccRCC and pRCC, chRCC exhibits a much-reduced microvessel density and a slower rate of glucose uptake, suggesting that chRCC cells favor a different mode of nutrient uptake to make up for the microenvironment’s lack of nutrients. In order to obtain extracellular macromolecules as a source of nutrition for cell survival and proliferation, chRCC cells can activate the endocytosis and downstream lysosomal pathways, which is demonstrated by the abundance increase in proteins implicated in these pathways and their enzymatic activity [207].
A limitation of the present research is that most of the discussed metabolic reprogramming refers to RCC subtypes because of the limited number of papers in the literature about the other types of renal cancer, taking into consideration that RCC represents the most common malignancy of the kidney. However, having a better understanding of the molecular events and signaling pathways involved in renal cancer could help improve the management and the tools used to diagnose, treat, and monitor renal cancer patients. The management of renal cancer is difficult in all aspects, from diagnosis to therapy and follow-up of the patients. Usually, the differential diagnosis of benign or malignant tumors is based on imaging and a biopsy. Nevertheless, these methods are not always efficient in renal cancer because of the heterogeneity of the tumors. Therefore, additional diagnosis and monitoring tools, such as artificial intelligence, genomics, and radiogenomics have been developed in recent years, which together with the greater knowledge of the molecular events associated with the histopathology aspects, could provide new perspectives into clinical practice [249][250].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24098391
References
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285.
- Clark, D.; Dhanasekaran, S.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.; Chang, H.C. Clinical, Proteomic Tumor Analysis. Cell 2019, 179, 964–983.
- Creighton, C.J. Proteomic signatures of clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 133–134.
- Irmisch, A.; Bonilla, X.; Chevrier, S.; Lehmann, K.-V.; Singer, F.; Toussaint, N.C.; Esposito, C.; Mena, J.; Milani, E.S.; Casanova, R. The Tumor Profiler Study: Integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell 2021, 39, 288–293.
- International Association of Cancer Registries (IACR); International Agency for Research on Cancer(IARC); World Health Organization(WHO). International Classification of Diseases for Oncology (ICD-O)—ICD-O-3.2; Updated 25 January 2021. Available online: http://www.iacr.com.fr/index.php?Itemid=577 (accessed on 15 January 2023).
- Zbar, B.; Brauch, H.; Talmadge, C.; Linehan, M. Loss of alleles of loci on the short arm of chromosome. Nature 1987, 3, 721–724.
- Mizutani, K.; Yokoi, S.; Sawada, S.; Sakamoto, I.; Kameyama, K.; Kamei, S.; Hirade, K.; Sugiyama, S.; Matsunaga, K.; Yamada, T. Derivative Chromosome 3 Loss from t (3; 6)(q12; q14) Followed by Differential VHL Mutations Underlie Multifocal ccRCC. Cancer Genom. Proteom. 2022, 19, 740–746.
- Bentz, M.; Bergerheim, U.; Li, C.; Joos, S.; Werner, C.; Baudis, M.; Gnarra, J.; Merino, M.; Zbar, B.; Linehan, W. Chromosome imbalances in papillary renal cell carcinoma and first cytogenetic data of familial cases analyzed by comparative genomic hybridization. Cytogenet. Genome Res. 1996, 75, 17–21.
- Testa, U.; Pelosi, E.; Castelli, G. Genetic alterations in renal cancers: Identification of the mechanisms underlying cancer initiation and progression and of therapeutic targets. Medicines 2020, 7, 44.
- Schmidt, L.; Duh, F.-M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73.
- Webster, B.R.; Gopal, N.; Ball, M.W. Tumorigenesis Mechanisms Found in Hereditary Renal Cell Carcinoma: A Review. Genes 2022, 13, 2122.
- Alaghehbandan, R.; Przybycin, C.G.; Verkarre, V.; Mehra, R. Chromophobe renal cell carcinoma: Novel molecular insights and clinicopathologic updates. Asian J. Urol. 2021, 9, 1–11.
- Büscheck, F.; Fraune, C.; Garmestani, S.; Simon, R.; Kluth, M.; Hube-Magg, C.; Ketterer, K.; Eichelberg, C.; Höflmayer, D.; Jacobsen, F. Y-chromosome loss is frequent in male renal tumors. Ann. Transl. Med. 2021, 9, 209.
- Kovacs, G.; Akhtar, M.; Beckwith, B.J.; Bugert, P.; Cooper, C.S.; Delahunt, B.; Eble, J.N.; Fleming, S.; Ljungberg, B.; Medeiros, L.J. The Heidelberg classification of renal cell tumours. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 1997, 183, 131–133.
- Suárez Rodríguez, C.; Marmolejo Castañeda, D.; Valdivia Bustamante, A.A.; Morales Barrera, R.; González Rodríguez, M.; Mateo Valderrama, J.; Semidey Raven, M.E.; Lorente García, D.; Trilla Herrera, E.; Carles Galceran, J. Update in collecting duct carcinoma: Current aspects of the clinical and molecular characterization of an orphan disease. Front. Oncol. 2022, 12, 970199.
- Muglia, V.F.; Prando, A. Carcinoma de células renais: Classificação histológica e correlação com métodos de imagem. Radiol. Bras. 2015, 48, 166–174.
- Campbell, M.T.; Jonasch, E.; Wood, C.G.; Tannir, N.M. Renal Cell Carcinoma. In The MD Anderson Manual of Medical Oncology, 3e; Kantarjian, H.M., Wolff, R.A., Eds.; McGraw-Hill Medical: New York, NY, USA, 2016.
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Garfield, K.; LaGrange, C.A. Renal cell cancer. In StatPearls ; StatPearls Publishing: Tampa, FL, USA, 2021.
- Nabi, S.; Kessler, E.R.; Bernard, B.; Flaig, T.W.; Lam, E.T. Renal cell carcinoma: A review of biology and pathophysiology. F1000Research 2018, 7, 307.
- Moch, H.; Amin, M.B.; Berney, D.M.; Compérat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R. The 2022 World Health Organization classification of tumours of the urinary system and male genital organs—Part A: Renal, penile, and testicular tumours. Eur. Urol. 2022, 82, 458–468.
- Chen, F.; Zhang, Y.; Şenbabaoğlu, Y.; Ciriello, G.; Yang, L.; Reznik, E.; Shuch, B.; Micevic, G.; De Velasco, G.; Shinbrot, E. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016, 14, 2476–2489.
- Li, Y.; Lih, T.-S.M.; Dhanasekaran, S.M.; Mannan, R.; Chen, L.; Cieslik, M.; Wu, Y.; Lu, R.J.-H.; Clark, D.J.; Kołodziejczak, I. Histopathologic and proteogenomic heterogeneity reveals features of clear cell renal cell carcinoma aggressiveness. Cancer Cell 2022, 41, 139–163.e17.
- Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; Vocke, C.D.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145.
- John, A.; Spain, L.; Hamid, A.A. Navigating the Current Landscape of Non-Clear Cell Renal Cell Carcinoma: A Review of the Literature. Curr. Oncol. 2023, 30, 923–937.
- Chen, Y.-B.; Xu, J.; Skanderup, A.J.; Dong, Y.; Brannon, A.R.; Wang, L.; Won, H.H.; Wang, P.I.; Nanjangud, G.J.; Jungbluth, A.A. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat. Commun. 2016, 7, 13131.
- Sharma, R.; Kannourakis, G.; Prithiviraj, P.; Ahmed, N. Precision medicine–an optimal approach to patient care in Renal Cell Carcinoma. Front. Med. 2022, 9, 1240.
- Jonasch, E.; Gao, J.; Rathmell, W.K. Renal cell carcinoma. BMJ 2014, 349, g4797.
- Mohd, A.B.; Ghannam, R.A.; Mohd, O.B.; Elayan, R.; Albakri, K.; Huneiti, N.; Daraghmeh, F.; Al-Khatatbeh, E.; Al-Thnaibat, M. Etiologies, Gross Appearance, Histopathological Patterns, Prognosis, and Best Treatments for Subtypes of Renal Carcinoma: An Educational Review. Cureus 2022, 14, e32338.
- Padala, S.A.; Kallam, A. Clear Cell Renal Carcinoma. In StatPearls; StatPearls Publishing. Copyright © 2023; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023.
- DeCastro, G.J.; McKiernan, J.M. Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol. Clin. N. Am. 2008, 35, 581–592.
- Novacescu, D.; Feciche, B.O.; Cumpanas, A.A.; Bardan, R.; Rusmir, A.V.; Bitar, Y.A.; Barbos, V.I.; Cut, T.G.; Raica, M.; Latcu, S.C. Contemporary Clinical Definitions, Differential Diagnosis, and Novel Predictive Tools for Renal Cell Carcinoma. Biomedicines 2022, 10, 2926.
- Dagher, J.; Delahunt, B.; Rioux-Leclercq, N.; Egevad, L.; Srigley, J.R.; Coughlin, G.; Dunglinson, N.; Gianduzzo, T.; Kua, B.; Malone, G. Clear cell renal cell carcinoma: Validation of World Health Organization/International Society of Urological Pathology grading. Histopathology 2017, 71, 918–925.
- Akhtar, M.; Al-Bozom, I.A.; Al Hussain, T. Papillary renal cell carcinoma (PRCC): An update. Adv. Anat. Pathol. 2019, 26, 124–132.
- Kim, J.-Y.; Jeong, H.-O.; Heo, D.S.; Keam, B.; Moon, K.C.; Kwak, C.; Jang, J.; Kim, S.; Kim, J.-I.; Lee, S. Treatment strategy for papillary renal cell carcinoma type 2: A case series of seven patients treated based on next generation sequencing data. Ann. Transl. Med. 2020, 8, 124–132.
- Murugan, P.; Jia, L.; Dinatale, R.G.; Assel, M.; Benfante, N.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Sarungbam, J.; Sirintrapun, S.J. Papillary renal cell carcinoma: A single institutional study of 199 cases addressing classification, clinicopathologic and molecular features, and treatment outcome. Mod. Pathol. 2022, 35, 825–835.
- Mendhiratta, N.; Muraki, P.; Sisk, A.E., Jr.; Shuch, B. Papillary renal cell carcinoma. In Proceedings of the Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2021; pp. 327–337.
- Tretiakova, M. Journal of Pathology and Translational Medicine. J. Pathol. Transl. Med. 2022, 56, 383–384.
- Roldan-Romero, J.M.; Santos, M.; Lanillos, J.; Caleiras, E.; Anguera, G.; Maroto, P.; García-Donas, J.; de Velasco, G.; Martinez-Montes, Á.M.; Calsina, B. Molecular characterization of chromophobe renal cell carcinoma reveals mTOR pathway alterations in patients with poor outcome. Mod. Pathol. 2020, 33, 2580–2590.
- Garje, R.; Elhag, D.; Yasin, H.A.; Acharya, L.; Vaena, D.; Dahmoush, L. Comprehensive review of chromophobe renal cell carcinoma. Crit. Rev. Oncol./Hematol. 2021, 160, 103287.
- Rysz, J.; Franczyk, B.; Ławiński, J.; Gluba-Brzózka, A. Characteristics of clear cell papillary renal cell carcinoma (ccpRCC). Int. J. Mol. Sci. 2021, 23, 151.
- Rohan, S.M.; Xiao, Y.; Liang, Y.; Dudas, M.E.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Reuter, V.E.; Rosenblum, M.K.; Russo, P. Clear-cell papillary renal cell carcinoma: Molecular and immunohistochemical analysis with emphasis on the von Hippel–Lindau gene and hypoxia-inducible factor pathway-related proteins. Mod. Pathol. 2011, 24, 1207–1220.
- Xu, J.; Reznik, E.; Lee, H.-J.; Gundem, G.; Jonsson, P.; Sarungbam, J.; Bialik, A.; Sanchez-Vega, F.; Creighton, C.J.; Hoekstra, J. Abnormal oxidative metabolism in a quiet genomic background underlies clear cell papillary renal cell carcinoma. eLife 2019, 8, e38986.
- Cabanillas, G.; Montoya-Cerrillo, D.; Kryvenko, O.N.; Pal, S.K.; Arias-Stella III, J.A. Collecting duct carcinoma of the kidney: Diagnosis and implications for management. In Proceedings of the Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2022; pp. 525–536.
- Gupta, S. An update on the pathology of collecting duct & papillary renal cell carcinoma with a discussion of SNP-Arrays as an emerging laboratory technique. In Proceedings of the Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2022; pp. 497–498.
- Msaouel, P.; Hong, A.L.; Mullen, E.A.; Atkins, M.B.; Walker, C.L.; Lee, C.-H.; Carden, M.A.; Genovese, G.; Linehan, W.M.; Rao, P. Updated recommendations on the diagnosis, management, and clinical trial eligibility criteria for patients with renal medullary carcinoma. Clin. Genitourin. Cancer 2019, 17, 1–6.
- Su, Y.; Hong, A.L. Recent Advances in Renal Medullary Carcinoma. Int. J. Mol. Sci. 2022, 23, 7097.
- Basher, F.; Dutcher, G.; England, J.S.; Lopes, G. Unusual Presentation of Renal Medullary Carcinoma with Undiagnosed Sickle Cell Trait. Cureus 2020, 12, e10731.
- Elliott, A.; Bruner, E. Renal medullary carcinoma. Arch. Pathol. Lab. Med. 2019, 143, 1556–1561.
- Kohashi, K.; Oda, Y. Oncogenic roles of SMARCB 1/INI 1 and its deficient tumors. Cancer Sci. 2017, 108, 547–552.
- Miller, K.E.; Wheeler, G.; LaHaye, S.; Schieffer, K.M.; Cearlock, S.; Venkata, L.P.R.; Bravo, A.O.; Grischow, O.E.; Kelly, B.J.; White, P. Molecular heterogeneity in pediatric malignant rhabdoid tumors in patients with multi-organ involvement. Front. Oncol. 2022, 12, 932337.
- Andeen, N.K.; Tretiakova, M. Kidney Tumor. Available online: https://www.pathologyoutlines.com/topic/kidneytumormalignantnos.html (accessed on 2 February 2023).
- Sirohi, D.; Smith, S.C.; Agarwal, N.; Maughan, B.L. Unclassified renal cell carcinoma: Diagnostic difficulties and treatment modalities. Res. Rep. Urol. 2018, 10, 205–217.
- Miyazaki, J.; Nishiyama, H. Epidemiology of urothelial carcinoma. Int. J. Urol. 2017, 24, 730–734.
- Tomiyama, E.; Fujita, K.; Hashimoto, M.; Adomi, S.; Kawashima, A.; Minami, T.; Yoshimura, K.; Uemura, H.; Nonomura, N. Comparison of molecular profiles of upper tract urothelial carcinoma vs. urinary bladder cancer in the era of targeted therapy: A narrative review. Transl. Androl. Urol. 2022, 11, 1747.
- Ogbue, O.; Haddad, A.; Almassi, N.; Lapinski, J.; Daw, H. Overview of histologic variants of urothelial carcinoma: Current trends and narrative review on treatment outcomes. Transl. Androl. Urol. 2022, 11, 877.
- Xing, X.; Yuan, X.; Liu, T.; Dai, M.; Fan, Y.; Liu, C.; Strååt, K.; Björkholm, M.; Xu, D. Regulatory region mutations of TERT, PLEKHS1 and GPR126 genes as urinary biomarkers in upper tract urothelial carcinomas. J. Cancer 2021, 12, 3853.
- Caldwell, B.T.; Wilcox, D.T.; Cost, N.G. Current management for pediatric urologic oncology. Adv. Pediatr. 2017, 64, 191–223.
- Nazemi, A.; Daneshmand, S.; Chang, A. Pediatric genitourinary tumors: Distribution, demographics, and outcomes. Pediatr. Investig. 2022, 6, 85–92.
- Available online: https://www.cancer.org/cancer/wilms-tumor/about/key-statistics.html (accessed on 12 March 2023).
- Dome, J.S.; Graf, N.; Geller, J.I.; Fernandez, C.V.; Mullen, E.A.; Spreafico, F.; Van den Heuvel-Eibrink, M.; Pritchard-Jones, K. Advances in Wilms tumor treatment and biology: Progress through international collaboration. J. Clin. Oncol. 2015, 33, 2999.
- Tian, X.-M.; Xiang, B.; Jin, L.-M.; Mi, T.; Wang, J.-K.; Zhanghuang, C.; Zhang, Z.-X.; Chen, M.-L.; Shi, Q.-L.; Liu, F. Immune-related gene signature associates with immune landscape and predicts prognosis accurately in patients with Wilms tumour. Front. Immunol. 2022, 13, 5230.
- Royer-Pokora, B.; Busch, M.A.; Tenbusch, S.; Schmidt, M.; Beier, M.; Woods, A.D.; Thiele, H.; Mora, J. Comprehensive biology and genetics compendium of wilms tumor cell lines with different wt1 mutations. Cancers 2020, 13, 60.
- Huff, V. Wilms’ tumours: About tumour suppressor genes, an oncogene and a chameleon gene. Nat. Rev. Cancer 2011, 11, 111–121.
- Sahu, D.K.; Singh, N.; Das, M.; Rawat, J.; Gupta, D.K. Differential expression profiling of onco and tumor-suppressor genes from major-signaling pathways in Wilms’ tumor. Pediatr. Surg. Int. 2022, 38, 1601–1617.
- Ruteshouser, E.C.; Robinson, S.M.; Huff, V. Wilms tumor genetics: Mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosom. Cancer 2008, 47, 461–470.
- Xie, W.; Wei, L.; Guo, J.; Guo, H.; Song, X.; Sheng, X. Physiological functions of Wilms’ tumor 1-associating protein and its role in tumourigenesis. J. Cell. Biochem. 2019, 120, 10884–10892.
- Lanzkowsky, P.; Lipton, J.; Fish, J.D. Lanzkowsky’s Manual of Pediatric Hematology and Oncology; Elsevier: Amsterdam, The Netherlands, 2016.
- Sebire, N.J.; Vujanic, G.M. Paediatric renal tumours: Recent developments, new entities and pathological features. Histopathology 2009, 54, 516–528.
- van der Beek, J.N.; Hol, J.A.; Coulomb-l’Hermine, A.; Graf, N.; van Tinteren, H.; Pritchard-Jones, K.; Houwing, M.E.; de Krijger, R.R.; Vujanic, G.M.; Dzhuma, K. Characteristics and outcome of pediatric renal cell carcinoma patients registered in the International Society of Pediatric Oncology (SIOP) 93-01, 2001 and UK-IMPORT database: A report of the SIOP-Renal Tumor Study Group. Int. J. Cancer 2021, 148, 2724–2735.
- Dome, J.S.; Cotton, C.A.; Perlman, E.J.; Breslow, N.E.; Kalapurakal, J.A.; Ritchey, M.L.; Grundy, P.E.; Malogolowkin, M.; Beckwith, J.B.; Shamberger, R.C. Treatment of anaplastic histology Wilms’ tumor: Results from the fifth National Wilms’ Tumor Study. J. Clin. Oncol. 2006, 24, 2352–2358.
- Chintagumpala, M.M.; Perlman, E.J.; Tornwall, B.; Chi, Y.Y.; Kim, Y.; Hoffer, F.A.; Kalapurakal, J.A.; Warwick, A.B.; Shamberger, R.C.; Khanna, G. Outcomes based on histopathologic response to preoperative chemotherapy in children with bilateral Wilms tumor: A prospective study (COG AREN0534). Cancer 2022, 128, 2493–2503.
- Daw, N.C.; Chi, Y.-Y.; Kalapurakal, J.A.; Kim, Y.; Hoffer, F.A.; Geller, J.I.; Perlman, E.J.; Ehrlich, P.F.; Mullen, E.A.; Warwick, A.B. Activity of vincristine and irinotecan in diffuse anaplastic Wilms tumor and therapy outcomes of stage II to IV disease: Results of the Children’s Oncology Group AREN0321 study. J. Clin. Oncol. 2020, 38, 1558.
- Groenendijk, A.; Spreafico, F.; de Krijger, R.R.; Drost, J.; Brok, J.; Perotti, D.; van Tinteren, H.; Venkatramani, R.; Godziński, J.; Rübe, C. Prognostic factors for Wilms tumor recurrence: A review of the literature. Cancers 2021, 13, 3142.
- Öztürk, H. Prognostic features of renal sarcomas. Oncol. Lett. 2015, 9, 1034–1038.
- Uhlig, J.; Uhlig, A.; Bachanek, S.; Onur, M.R.; Kinner, S.; Geisel, D.; Köhler, M.; Preibsch, H.; Puesken, M.; Schramm, D. Primary renal sarcomas: Imaging features and discrimination from non-sarcoma renal tumors. Eur. Radiol. 2022, 32, 981–989.
- Chaudhary, D.; Rath, A.; Mandal, S.; Khurana, N.; Agarwal, P. Primary leiomyosarcoma kidney–A rare entity with a diagnostic challenge. J. Cancer Res. Ther. 2022, 18, 1186–1188.
- Li, T.; Huang, S.; Yan, W.; Zhang, Y.; Guo, Q. FOXF2 Regulates PRUNE2 Transcription in the Pathogenesis of Colorectal Cancer. Technol. Cancer Res. Treat. 2022, 21, 15330338221118717.
- Badeloe, S.; Van Geest, A.J.; Van Marion, A.M.; Frank, J. Absence of fumarate hydratase mutation in a family with cutaneous leiomyosarcoma and renal cancer. Int. J. Dermatol. 2008, 47, 18–20.
- Yagi, Y.; Abeto, N.; Shiraishi, J.; Miyata, C.; Inoue, S.; Murakami, H.; Nakashima, M.; Sugano, K.; Ushiama, M.; Yoshida, T. A novel pathogenic variant of the FH gene in a family with hereditary leiomyomatosis and renal cell carcinoma. Hum. Genome Var. 2022, 9, 3.
- Darlington, D.; Anitha, F.S. Atypical presentation of renal leiomyosarcoma: A case report. Cureus 2019, 11.
- Silveira, S.M.; Villacis, R.A.R.; Marchi, F.A.; Barros Filho, M.d.C.; Drigo, S.A.; Neto, C.S.; Lopes, A.; da Cunha, I.W.; Rogatto, S.R. Genomic signatures predict poor outcome in undifferentiated pleomorphic sarcomas and leiomyosarcomas. PLoS ONE 2013, 8, e67643.
- Roulleaux Dugage, M.; Nassif, E.F.; Italiano, A.; Bahleda, R. Improving immunotherapy efficacy in soft-tissue sarcomas: A biomarker driven and histotype tailored review. Front. Immunol. 2021, 12, 5237.
- Gonzalez, C.G.; Akula, S.; Burleson, M. The role of mediator subunit 12 in tumorigenesis and cancer therapeutics. Oncol. Lett. 2022, 23, 74.
- Rubisz, P.; Ciebiera, M.; Hirnle, L.; Zgliczyńska, M.; Łoziński, T.; Dzięgiel, P.; Kobierzycki, C. The Usefulness of Immunohistochemistry in the Differential Diagnosis of Lesions Originating from the Myometrium. Int. J. Mol. Sci. 2019, 20, 1136.
- Djiwa, T.; Sabi, K.A.; Simgban, P.; Bombonne, M.; Sama, B.M.; Tchaou, M.; Darré, T. Renal Leiomyosarcoma, a Rare Presentation. J. Kidney Cancer VHL 2022, 9, 51.
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. BioSystems 2015, 11, 1946–1954.
- Aoki, M.; Fujishita, T. Oncogenic roles of the PI3K/AKT/mTOR axis. Viruses Genes Cancer 2017, 407, 153–189.
- Polivka Jr, J.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175.
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657.
- Hawkins, P.; Stephens, L. PI3K signalling in inflammation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 882–897.
- Denley, A.; Kang, S.; Karst, U.; Vogt, P. Oncogenic signaling of class I PI3K isoforms. Oncogene 2008, 27, 2561–2574.
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635.
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425.
- Fry, M.J. Phosphoinositide 3-kinase signalling in breast cancer: How big a role might it play? Breast Cancer Res. 2001, 3, 304–312.
- Bénistant, C.; Chapuis, H.; Roche, S. A specific function for phosphatidylinositol 3-kinase α (p85α-p110α) in cell survival and for phosphatidylinositol 3-kinase β (p85α-p110β) in de novo DNA synthesis of human colon carcinoma cells. Oncogene 2000, 19, 5083–5090.
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase γ. Cell 2000, 103, 931–944.
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675.
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling pathways in inflammation and anti-inflammatory therapies. Curr. Pharm. Des. 2018, 24, 1449–1484.
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501.
- Hawkins, P.; Anderson, K.; Davidson, K.; Stephens, L. Signalling through Class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006, 34, 647–662.
- Andrews, S.; Stephens, L.R.; Hawkins, P.T. PI3K class IB pathway. Sci. STKE 2007, 2007, cm2.
- Wang, H.; Lo, W.-T.; Žagar, A.V.; Gulluni, F.; Lehmann, M.; Scapozza, L.; Haucke, V.; Vadas, O. Autoregulation of class II alpha PI3K activity by its lipid-binding PX-C2 domain module. Mol. Cell 2018, 71, 343–351.e4.
- Aung, K.T.; Yoshioka, K.; Aki, S.; Ishimaru, K.; Takuwa, N.; Takuwa, Y. The class II phosphoinositide 3-kinases PI3K-C2α and PI3K-C2β differentially regulate clathrin-dependent pinocytosis in human vascular endothelial cells. J. Physiol. Sci. 2019, 69, 263–280.
- Cisse, O.; Quraishi, M.; Gulluni, F.; Guffanti, F.; Mavrommati, I.; Suthanthirakumaran, M.; Oh, L.C.; Schlatter, J.N.; Sarvananthan, A.; Broggini, M. Downregulation of class II phosphoinositide 3-kinase PI3K-C2β delays cell division and potentiates the effect of docetaxel on cancer cell growth. J. Exp. Clin. Cancer Res. 2019, 38, 472.
- Chikh, A.; Ferro, R.; Abbott, J.J.; Piñeiro, R.; Buus, R.; Iezzi, M.; Ricci, F.; Bergamaschi, D.; Ostano, P.; Chiorino, G. Class II phosphoinositide 3-kinase C2β regulates a novel signaling pathway involved in breast cancer progression. Oncotarget 2016, 7, 18325.
- Liu, P.; Morrison, C.; Wang, L.; Xiong, D.; Vedell, P.; Cui, P.; Hua, X.; Ding, F.; Lu, Y.; James, M. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 2012, 33, 1270–1276.
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in cancer: Any good news? Front. Oncol. 2013, 3, 108.
- Wurmser, A.E.; Gary, J.D.; Emr, S.D. Phosphoinositide 3-kinases and their FYVE domain-containing effectors as regulators of vacuolar/lysosomal membrane trafficking pathways. J. Biol. Chem. 1999, 274, 9129–9132.
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3–kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530.
- Roggo, L.; Bernard, V.; Kovacs, A.L.; Rose, A.M.; Savoy, F.; Zetka, M.; Wymann, M.P.; Müller, F. Membrane transport in Caenorhabditis elegans: An essential role for VPS34 at the nuclear membrane. EMBO J. 2002, 21, 1673–1683.
- Burda, P.; Padilla, S.M.; Sarkar, S.; Emr, S.D. Retromer function in endosome-to-Golgi retrograde transport is regulated by the yeast Vps34 PtdIns 3-kinase. J. Cell Sci. 2002, 115, 3889–3900.
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271.
- Markman, B.; Dienstmann, R.; Tabernero, J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget 2010, 1, 530.
- Matheny Jr, R.W.; Adamo, M.L. Current perspectives on Akt Akt-ivation and Akt-ions. Exp. Biol. Med. 2009, 234, 1264–1270.
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28.
- Testa, J.R.; Bellacosa, A. AKT plays a central role in tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985.
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw III, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731.
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31.
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Santaliz Casiano, A.; Wrobel, K.; Rossi, G.; Smith, R.L. Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR SignalingERα in Obesity-Associated Breast Cancer. Cancer Res. 2019, 79, 2494–2510.
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22.
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574.
- Hall, M. mTOR—What does it do? In Proceedings of the Transplantation Proceedings; Elsevier: Amsterdam, The Netherlands, 2008; pp. S5–S8.
- Sangüesa, G.; Roglans, N.; Baena, M.; Velázquez, A.M.; Laguna, J.C.; Alegret, M. mTOR is a key protein involved in the metabolic effects of simple sugars. Int. J. Mol. Sci. 2019, 20, 1117.
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594.
- Ruan, C.; Ouyang, X.; Liu, H.; Li, S.; Jin, J.; Tang, W.; Xia, Y.; Su, B. Sin1-mediated mTOR signaling in cell growth, metabolism and immune response. Natl. Sci. Rev. 2019, 6, 1149–1162.
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302.
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316.
- Deng, F.; Ma, Y.-X.; Liang, L.; Zhang, P.; Feng, J. The pro-apoptosis effect of sinomenine in renal carcinoma via inducing autophagy through inactivating PI3K/AKT/mTOR pathway. Biomed. Pharmacother. 2018, 97, 1269–1274.
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117.
- Martínez-Sáez, O.; Borau, P.G.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Grande, E. Targeting HIF-2 α in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit. Rev. Oncol./Hematol. 2017, 111, 117–123.
- Choueiri, T.K.; Kaelin Jr, W.G. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530.
- Baba, M.; Hirai, S.; Yamada-Okabe, H.; Hamada, K.; Tabuchi, H.; Kobayashi, K.; Kondo, K.; Yoshida, M.; Yamashita, A.; Kishida, T. Loss of von Hippel-Lindau protein causes cell density dependent deregulation of CyclinD1 expression through hypoxia-inducible factor. Oncogene 2003, 22, 2728–2738.
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216.
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur. Urol. 2016, 69, 646–657.
- Koul, H.; Huh, J.-S.; Rove, K.O.; Crompton, L.; Koul, S.; Meacham, R.B.; Kim, F.J. Molecular aspects of renal cell carcinoma: A review. Am. J. Cancer Res. 2011, 1, 240.
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156.
- Baldewijns, M.M.; van Vlodrop, I.J.; Vermeulen, P.B.; Soetekouw, P.M.; van Engeland, M.; de Bruïne, A.P. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 2010, 221, 125–138.
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576.
- Chen, Y.; Zhou, X. Research progress of mTOR inhibitors. Eur. J. Med. Chem. 2020, 208, 112820.
- Que, W.-C.; Qiu, H.-Q.; Cheng, Y.; Liu, M.-B.; Wu, C.-Y. PTEN in kidney cancer: A review and meta-analysis. Clin. Chim. Acta 2018, 480, 92–98.
- Brenner, W.; Färber, G.; Herget, T.; Lehr, H.A.; Hengstler, J.G.; Thüroff, J.W. Loss of tumor suppressor protein PTEN during renal carcinogenesis. Int. J. Cancer 2002, 99, 53–57.
- Hager, M.; Haufe, H.; Kemmerling, R.; Mikuz, G.; Kolbitsch, C.; Moser, P.L. PTEN expression in renal cell carcinoma and oncocytoma and prognosis. Pathology 2007, 39, 482–485.
- Owonikoko, T.K.; Khuri, F.R. Targeting the PI3K/AKT/mTOR pathway: Biomarkers of success and tribulation. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, e395–e401.
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; Jiang, Z.; Oliva, E.; Jozwiak, S.; Nussbaum, R.L. Renal cell carcinoma in tuberous sclerosis complex. Am. J. Surg. Pathol. 2014, 38, 895.
- Gargalionis, A.N.; Sarlani, E.; Stofas, A.; Malakou, L.S.; Adamopoulos, C.; Bamias, A.; Boutati, E.; Constantinides, C.A.; Stravodimos, K.G.; Piperi, C. Polycystin-1 induces activation of the PI3K/AKT/mTOR pathway and promotes angiogenesis in renal cell carcinoma. Cancer Lett. 2020, 489, 135–143.
- Henske, E.P.; Cornejo, K.M.; Wu, C.-L. Renal cell carcinoma in tuberous sclerosis complex. Genes 2021, 12, 1585.
- da Costa E Silva, V.T.; Costalonga, E.C.; Coelho, F.O.; Caires, R.A.; Burdmann, E.A. Assessment of kidney function in patients with cancer. Adv. Chronic Kidney Dis. 2018, 25, 49–56.
- Orillion, A.; Damayanti, N.P.; Shen, L.; Adelaiye-Ogala, R.; Affronti, H.; Elbanna, M.; Chintala, S.; Ciesielski, M.; Fontana, L.; Kao, C. Dietary protein restriction reprograms tumor-associated macrophages and enhances immunotherapy. Clin. Cancer Res. 2018, 24, 6383–6395.
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin–PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283.
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1.
- Massari, F.; Ciccarese, C.; Santoni, M.; Brunelli, M.; Piva, F.; Modena, A.; Bimbatti, D.; Fantinel, E.; Santini, D.; Cheng, L. Metabolic alterations in renal cell carcinoma. Cancer Treat. Rev. 2015, 41, 767–776.
- Becattini, B.; Marone, R.; Zani, F.; Arsenijevic, D.; Seydoux, J.; Montani, J.-P.; Dulloo, A.G.; Thorens, B.; Preitner, F.; Wymann, M.P. PI3Kγ within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, E854–E863.
- Wettersten, H.I. Reprogramming of metabolism in kidney cancer. In Proceedings of the Seminars in Nephrology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 2–13.
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486.
- Wettersten, H.I.; Aboud, O.A.; Lara Jr, P.N.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419.
- Ku, B.M.; Lee, C.-H.; Lee, S.-H.; Kim, S.-Y. Increased expression of transglutaminase 2 drives glycolytic metabolism in renal carcinoma cells. Amino Acids 2014, 46, 1527–1536.
- Corrales, P.; Izquierdo-Lahuerta, A.; Medina-Gómez, G. Maintenance of kidney metabolic homeostasis by PPAR gamma. Int. J. Mol. Sci. 2018, 19, 2063.
- Cheng, L.; Ge, M.; Lan, Z.; Ma, Z.; Chi, W.; Kuang, W.; Sun, K.; Zhao, X.; Liu, Y.; Feng, Y. Zoledronate dysregulates fatty acid metabolism in renal tubular epithelial cells to induce nephrotoxicity. Arch. Toxicol. 2018, 92, 469–485.
- Myszczyszyn, A.; Czarnecka, A.M.; Matak, D.; Szymanski, L.; Lian, F.; Kornakiewicz, A.; Bartnik, E.; Kukwa, W.; Kieda, C.; Szczylik, C. The role of hypoxia and cancer stem cells in renal cell carcinoma pathogenesis. Stem Cell Rev. Rep. 2015, 11, 919–943.
- Ghazi, S.; Polesel, M.; Hall, A.M. Targeting glycolysis in proliferative kidney diseases. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1531–F1535.
- Du, C.; Zhu, Y.; Yang, Y.; Mu, L.; Yan, X.; Wu, M.; Zhou, C.; Wu, H.; Zhang, W.; Wu, Y. C1q/tumour necrosis factor-related protein-3 alleviates high-glucose-induced lipid accumulation and necroinflammation in renal tubular cells by activating the adenosine monophosphate-activated protein kinase pathway. Int. J. Biochem. Cell Biol. 2022, 149, 106247.
- Hoerner, C.R.; Miao, S.Y.; Hsieh, J.J.; Fan, A.C. Targeting Metabolic Pathways in Kidney Cancer: Rationale and Therapeutic Opportunities. Cancer J. 2020, 26, 407–418.
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Baltazar, F.; Jerónimo, C. The metabolic landscape of urological cancers: New therapeutic perspectives. Cancer Lett. 2020, 477, 76–87.
- Nakaigawa, N.; Kondo, K.; Ueno, D.; Namura, K.; Makiyama, K.; Kobayashi, K.; Shioi, K.; Ikeda, I.; Kishida, T.; Kaneta, T. The acceleration of glucose accumulation in renal cell carcinoma assessed by FDG PET/CT demonstrated acquisition of resistance to tyrosine kinase inhibitor therapy. BMC Cancer 2017, 17, 39.
- Kuroda, N.; Ohe, C.; Kawakami, F.; Mikami, S.; Furuya, M.; Matsuura, K.; Moriyama, M.; Nagashima, Y.; Zhou, M.; Petersson, F. Clear cell papillary renal cell carcinoma: A review. Int. J. Clin. Exp. Pathol. 2014, 7, 7312.
- Chakraborty, S.; Balan, M.; Sabarwal, A.; Choueiri, T.K.; Pal, S. Metabolic reprogramming in renal cancer: Events of a metabolic disease. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188559.
- Reustle, A.; Menig, L.S.; Leuthold, P.; Hofmann, U.; Stühler, V.; Schmees, C.; Becker, M.; Haag, M.; Klumpp, V.; Winter, S. Nicotinamide-N-methyltransferase is a promising metabolic drug target for primary and metastatic clear cell renal cell carcinoma. Clin. Transl. Med. 2022, 12, e883.
- Xiao, Y.; Rabien, A.; Buschow, R.; Amtislavskiy, V.; Busch, J.; Kilic, E.; Villegas, S.L.; Timmermann, B.; Schütte, M.; Mielke, T. Endocytosis-Mediated Replenishment of Amino Acids Favors Cancer Cell Proliferation and Survival in Chromophobe Renal Cell CarcinomaEndocytosis Activation in Chromophobe Renal Cell Carcinoma. Cancer Res. 2020, 80, 5491–5501.
- Ciccarese, C.; Santoni, M.; Massari, F.; Modena, A.; Piva, F.; Conti, A.; Mazzucchelli, R.; Cheng, L.; Lopez-Beltran, A.; Scarpelli, M. Metabolic alterations in renal and prostate cancer. Curr. Drug Metab. 2016, 17, 150–155.
- van der Mijn, J.C.; Panka, D.J.; Geissler, A.K.; Verheul, H.; Mier, J.W. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. 2016, 4, 14.
- Li, F.; Aljahdali, I.A.; Zhang, R.; Nastiuk, K.L.; Krolewski, J.J.; Ling, X. Kidney cancer biomarkers and targets for therapeutics: Survivin (BIRC5), XIAP, MCL-1, HIF1α, HIF2α, NRF2, MDM2, MDM4, p53, KRAS and AKT in renal cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 254.
- Liu, Q.; Galli, S.; Srinivasan, R.; Linehan, W.M.; Tsokos, M.; Merino, M.J. Renal medullary carcinoma: Molecular, immunohistochemistry, and morphologic correlation. Am. J. Surg. Pathol. 2013, 37, 368.
- Hervouet, E.; Simonnet, H.; Godinot, C. Mitochondria and reactive oxygen species in renal cancer. Biochimie 2007, 89, 1080–1088.
- Miikkulainen, P.; Högel, H.; Rantanen, K.; Suomi, T.; Kouvonen, P.; Elo, L.L.; Jaakkola, P.M. HIF prolyl hydroxylase PHD3 regulates translational machinery and glucose metabolism in clear cell renal cell carcinoma. Cancer Metab. 2017, 5, 5.
- Leisz, S.; Schulz, K.; Erb, S.; Oefner, P.; Dettmer, K.; Mougiakakos, D.; Wang, E.; Marincola, F.M.; Stehle, F.; Seliger, B. Distinct von Hippel-Lindau gene and hypoxia-regulated alterations in gene and protein expression patterns of renal cell carcinoma and their effects on metabolism. Oncotarget 2015, 6, 11395.
- Haase, V.H. Renal cancer: Oxygen meets metabolism. Exp. Cell Res. 2012, 318, 1057–1067.
- Bratslavsky, G.; Sudarshan, S.; Neckers, L.; Linehan, W.M. Pseudohypoxic pathways in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 4667–4671.
- Sanchez, D.J.; Simon, M.C. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2018, 1870, 23–31.
- Dizman, N.; Philip, E.J.; Pal, S.K. Genomic profiling in renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 435–451.
- Guo, X.; Zhang, Q. The emerging role of histone demethylases in renal cell carcinoma. J. Kidney Cancer VHL 2017, 4, 1–5.
- Shuch, B.; Linehan, W.M.; Srinivasan, R. Aerobic glycolysis: A novel target in kidney cancer. Expert Rev. Anticancer Ther. 2013, 13, 711–719.
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget 2015, 6, 13371.
- Takai, T.; Tsujino, T.; Yoshikawa, Y.; Inamoto, T.; Sugito, N.; Kuranaga, Y.; Heishima, K.; Soga, T.; Hayashi, K.; Miyata, K. Synthetic miR-143 exhibited an anti-cancer effect via the downregulation of K-RAS networks of renal cell cancer cells in vitro and in vivo. Mol. Ther. 2019, 27, 1017–1027.
- Liu, Y.; Meng, L.-L.; Li, J.-W.; Jin, Y.-S.; An, R.-H. A Randomized Study on the Effect of Metformin Combined with Intensive-Exercise Diet Therapy on Glucose and Lipid Metabolism and Islet Function in Patients with Renal Cell Carcinoma and Diabetes. Dis. Mrk. 2022, 2022, 7383745.
- Kobayashi, M.; Uematsu, T.; Tokura, Y.; Takei, K.; Sakamoto, K.; Narimatsu, T.; Nukui, A.; Kamai, T. Immunohistochemical expressionof sodium-dependent glucose transporter-2 (SGLT-2) in clear cell renal carcinoma: Possible prognostic implications. Int. Braz. J. Urol. 2019, 45, 169–178.
- Godinot, C.; de Laplanche, E.; Hervouet, E.; Simonnet, H. Actuality of Warburg’s views in our understanding of renal cancer metabolism. J. Bioenerg. Biomembr. 2007, 39, 235–241.
- Kaelin, W.G. Von Hippel–Lindau disease: Insights into oxygen sensing, protein degradation, and cancer. J. Clin. Investig. 2022, 132, e162480.
- Gu, Y.-R.; Kim, J.; Na, J.C.; Han, W.K. Mitochondrial metabolic reprogramming by SIRT3 regulation ameliorates drug resistance in renal cell carcinoma. PLoS ONE 2022, 17, e0269432.
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288.
- Lai, Y.; Tang, F.; Huang, Y.; He, C.; Chen, C.; Zhao, J.; Wu, W.; He, Z. The tumour microenvironment and metabolism in renal cell carcinoma targeted or immune therapy. J. Cell. Physiol. 2021, 236, 1616–1627.
- Schaeffeler, E.; Büttner, F.; Reustle, A.; Klumpp, V.; Winter, S.; Rausch, S.; Fisel, P.; Hennenlotter, J.; Kruck, S.; Stenzl, A. Metabolic and lipidomic reprogramming in renal cell carcinoma subtypes reflects regions of tumor origin. Eur. Urol. Focus 2019, 5, 608–618.
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369.
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388.
- Felipe-Abrio, B.; Verdugo-Sivianes, E.M.; Carnero, A. c-MYB-and PGC1a-dependent metabolic switch induced by MYBBP1A loss in renal cancer. Mol. Oncol. 2019, 13, 1519–1533.
- Dey, P.; Son, J.Y.; Kundu, A.; Kim, K.S.; Lee, Y.; Yoon, K.; Yoon, S.; Lee, B.M.; Nam, K.T.; Kim, H.S. Knockdown of pyruvate kinase M2 inhibits cell proliferation, metabolism, and migration in renal cell carcinoma. Int. J. Mol. Sci. 2019, 20, 5622.
- Morais, M.; Dias, F.; Nogueira, I.; Leão, A.; Gonçalves, N.; Araújo, L.; Granja, S.; Baltazar, F.; Teixeira, A.L.; Medeiros, R. Cancer cells’ metabolism dynamics in renal cell carcinoma patients’ outcome: Influence of GLUT-1-Related hsa-miR-144 and hsa-miR-186. Cancers 2021, 13, 1733.
- Lu, Q.; Yan, S.; Sun, H.; Wang, W.; Li, Y.; Yang, X.; Jiang, X.; Che, Y.; Xi, Z. Akt inhibition attenuates rasfonin-induced autophagy and apoptosis through the glycolytic pathway in renal cancer cells. Cell Death Dis. 2015, 6, e2005.
- Gonçalves, E.; Sciacovelli, M.; Costa, A.S.; Tran, M.G.B.; Johnson, T.I.; Machado, D.; Frezza, C.; Saez-Rodriguez, J. Post-translational regulation of metabolism in fumarate hydratase deficient cancer cells. Metab. Eng. 2018, 45, 149–157.
- Ge, X.; Li, M.; Yin, J.; Shi, Z.; Fu, Y.; Zhao, N.; Chen, H.; Meng, L.; Li, X.; Hu, Z. Fumarate inhibits PTEN to promote tumorigenesis and therapeutic resistance of type2 papillary renal cell carcinoma. Mol. Cell 2022, 82, 1249–1260.e1247.
- Wang, Q.; Zhang, Y.; Zhang, B.; Fu, Y.; Zhao, X.; Zhang, J.; Zuo, K.; Xing, Y.; Jiang, S.; Qin, Z. Single-cell chromatin accessibility landscape in kidney identifies additional cell-of-origin in heterogenous papillary renal cell carcinoma. Nat. Commun. 2022, 13, 31.
- Linehan, W.M.; Rathmell, W.K. Kidney cancer. In Proceedings of the Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2012; pp. 948–951.
- Sudarshan, S.; Linehan, W.; Neckers, L. HIF and fumarate hydratase in renal cancer. Br. J. Cancer 2007, 96, 403–407.
- Giubellino, A.; Linehan, W.M.; Bottaro, D.P. Targeting the Met signaling pathway in renal cancer. Expert Rev. Anticancer Ther. 2009, 9, 785–793.
- Qi, X.; Li, Q.; Che, X.; Wang, Q.; Wu, G. The uniqueness of clear cell renal cell carcinoma: Summary of the process and abnormality of glucose metabolism and lipid metabolism in ccRCC. Front. Oncol. 2021, 11, 727778.
- Xiao, Y.; Meierhofer, D. Glutathione metabolism in renal cell carcinoma progression and implications for therapies. Int. J. Mol. Sci. 2019, 20, 3672.
- Bacigalupa, Z.A.; Rathmell, W.K. Beyond glycolysis: Hypoxia signaling as a master regulator of alternative metabolic pathways and the implications in clear cell renal cell carcinoma. Cancer Lett. 2020, 489, 19–28.
- Linehan, W.M.; Rouault, T.A. Molecular pathways: Fumarate hydratase-deficient kidney cancer—Targeting the Warburg effect in cancer. Clin. Cancer Res. 2013, 19, 3345–3352.
- Chowdhury, B.; Porter, E.G.; Stewart, J.C.; Ferreira, C.R.; Schipma, M.J.; Dykhuizen, E.C. PBRM1 regulates the expression of genes involved in metabolism and cell adhesion in renal clear cell carcinoma. PLoS ONE 2016, 11, e0153718.
- Xie, H.; Song, J.; Godfrey, J.; Riscal, R.; Skuli, N.; Nissim, I.; Simon, M.C. Glycogen metabolism is dispensable for tumour progression in clear cell renal cell carcinoma. Nat. Metab. 2021, 3, 327–336.
- Zheng, Q.; Li, P.; Zhou, X.; Qiang, Y.; Fan, J.; Lin, Y.; Chen, Y.; Guo, J.; Wang, F.; Xue, H. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics 2021, 11, 8674.
- Pal, K.; Cao, Y.; Gaisina, I.N.; Bhattacharya, S.; Dutta, S.K.; Wang, E.; Gunosewoyo, H.; Kozikowski, A.P.; Billadeau, D.D.; Mukhopadhyay, D. Inhibition of GSK-3 Induces Differentiation and Impaired Glucose Metabolism in Renal CancerGSK-3 Inhibition in Renal Cancer. Mol. Cancer Ther. 2014, 13, 285–296.
- Cougnon, M.; Carcy, R.; Melis, N.; Rubera, I.; Duranton, C.; Dumas, K.; Tanti, J.-F.; Pons, C.; Soubeiran, N.; Shkreli, M. Inhibition of eIF5A hypusination reprogrammes metabolism and glucose handling in mouse kidney. Cell Death Dis. 2021, 12, 283.
- Lameirinhas, A.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C. The complex interplay between metabolic reprogramming and epigenetic alterations in renal cell carcinoma. Genes 2019, 10, 264.
- Heravi, G.; Yazdanpanah, O.; Podgorski, I.; Matherly, L.H.; Liu, W. Lipid metabolism reprogramming in renal cell carcinoma. Cancer Metastasis Rev. 2022, 41, 17–31.
- Bobulescu, I.A.; Pop, L.M.; Mani, C.; Turner, K.; Rivera, C.; Khatoon, S.; Kairamkonda, S.; Hannan, R.; Palle, K. Renal lipid metabolism abnormalities in obesity and clear cell renal cell carcinoma. Metabolites 2021, 11, 608.
- Weiss, R.H. Metabolomics and metabolic reprogramming in kidney cancer. In Proceedings of the Seminars in Nephrology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 175–182.
- Hu, T.; Chen, X.; Lu, S.; Zeng, H.; Guo, L.; Han, Y. Biological Role and Mechanism of Lipid Metabolism Reprogramming Related Gene ECHS1 in Cancer. Technol. Cancer Res. Treat. 2022, 21, 15330338221140655.
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E. Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524.
- Triantafyllou, E.-A.; Georgatsou, E.; Mylonis, I.; Simos, G.; Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 1142–1152.
- Młynarczyk, G.; Mikłosz, A.; Suchański, J.; Reza, S.; Romanowicz, L.; Sobolewski, K.; Chabowski, A.; Baranowski, M. Grade-dependent changes in sphingolipid metabolism in clear cell renal cell carcinoma. J. Cell. Biochem. 2022, 123, 819–829.
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A. Integration of lipidomics and transcriptomics reveals reprogramming of the lipid metabolism and composition in clear cell renal cell carcinoma. Metabolites 2020, 10, 509.
- Li, X.; Liu, Z.; Xia, C.; Yan, K.; Fang, Z.; Fan, Y. SETD8 stabilized by USP17 epigenetically activates SREBP1 pathway to drive lipogenesis and oncogenesis of ccRCC. Cancer Lett. 2022, 527, 150–163.
- Pressly, J.D.; Gurumani, M.Z.; Varona Santos, J.T.; Fornoni, A.; Merscher, S.; Al-Ali, H. Adaptive and maladaptive roles of lipid droplets in health and disease. Am. J. Physiol.-Cell Physiol. 2022, 322, C468–C481.
- Li, W.; Wang, X.; Zhang, X.; Gong, P.; Ding, D.; Wang, N.; Wang, Z. Revealing potential lipid biomarkers in clear cell renal cell carcinoma using targeted quantitative lipidomics. Lipids Health Dis. 2021, 20, 160.
- Li, K.; Zhu, Y.; Cheng, J.; Liu, Y.; Yang, X.; Huang, H.; Peng, Z.; Xu, H. A Novel Lipid Metabolism Genes Signature for Clear Cell Renal Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Cell Dev. Biol. 2023, 11, 80.
- Aminzadeh, S.; Vidali, S.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Energy metabolism in neuroblastoma and Wilms tumor. Transl. Pediatr. 2015, 4, 20.
- Zhao, Z.; Lu, J.; Han, L.; Wang, X.; Man, Q.; Liu, S. Prognostic significance of two lipid metabolism enzymes, HADHA and ACAT2, in clear cell renal cell carcinoma. Tumor Biol. 2016, 37, 8121–8130.
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769.
- Tang, X.; Wu, J.; Ding, C.-K.; Lu, M.; Keenan, M.M.; Lin, C.-C.; Lin, C.-A.; Wang, C.C.; George, D.; Hsu, D.S. Cystine deprivation triggers programmed necrosis in VHL-deficient renal cell carcinomas. Cancer Res. 2016, 76, 1892–1903.
- Yang, H.; Zhang, X.; Liu, F.; Fan, J.; Wang, B.; Dong, C. SREBP1-driven lipid desaturation supports clear cell renal cell carcinoma growth through regulation of NF-κB signaling. Biochem. Biophys. Res. Commun. 2018, 495, 1383–1388.
- Yu, W.; Yang, X.; Zhang, Q.; Sun, L.; Yuan, S.; Xin, Y. Targeting GLS1 to cancer therapy through glutamine metabolism. Clin. Transl. Oncol. 2021, 23, 2253–2268.
- Hoerner, C.R.; Chen, V.J.; Fan, A.C. The ‘Achilles Heel’of metabolism in renal cell carcinoma: Glutaminase inhibition as a rational treatment strategy. Kidney Cancer 2019, 3, 15–29.
- Tong, Y.; Kai, J.; Wang, S.; Yu, Y.; Xie, S.; Zheng, H.; Wang, Y.; Liu, Y.; Zhu, K.; Guan, X.; et al. VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1α/HO-1 pathway. Cell Death Dis. 2021, 12, 621.
- Zhang, H.; Yu, L.; Chen, J.; Liu, L.; Yang, X.; Cui, H.; Yue, G. Role of metabolic reprogramming of long non-coding RNA in clear cell renal cell carcinoma. J. Cancer 2022, 13, 691.
- Li, Z.; Wang, Y.; Wu, H.; Zhang, L.; Yang, P.; Li, Z. GRP78 enhances the glutamine metabolism to support cell survival from glucose deficiency by modulating the β-catenin signaling. Oncotarget 2014, 5, 5369.
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G. Glutaminase and poly (ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645.
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Pérez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; López-Larrubia, P. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385.
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292.
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40.
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451.
- Wang, J.; Chang, H.; Su, M.; Qiao, Y.; Sun, H.; Zhao, Y.; Zhang, S.; Shan, C. Identification of HGD and GSTZ1 as Biomarkers Involved Metabolic Reprogramming in Kidney Renal Clear Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 4583.
- Trott, J.F.; Hwang, V.J.; Ishimaru, T.; Chmiel, K.J.; Zhou, J.X.; Shim, K.; Stewart, B.J.; Mahjoub, M.R.; Jen, K.-Y.; Barupal, D.K. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am. J. Physiol.-Ren. Physiol. 2018, 315, F1855–F1868.
- Busnatu, Ș.-S.; Andronic, O.; Pană, M.-A.; Stoian, A.P.; Scafa-Udriște, A.; Păun, N.; Stanciu, S. Oral Arginine Supplementation in Healthy Individuals Performing Regular Resistance Training. In Proceedings of the Healthcare; SAGE: Thousand Oaks, CA, USA, 2023; p. 182.
- Hulea, L.; Gravel, S.-P.; Morita, M.; Cargnello, M.; Uchenunu, O.; Im, Y.K.; Lehuédé, C.; Ma, E.H.; Leibovitch, M.; McLaughlan, S. Translational and HIF-1α-dependent metabolic reprogramming underpin metabolic plasticity and responses to kinase inhibitors and biguanides. Cell Metab. 2018, 28, 817–832.e8.
- Miess, H.; Dankworth, B.; Gouw, A.M.; Rosenfeldt, M.; Schmitz, W.; Jiang, M.; Saunders, B.; Howell, M.; Downward, J.; Felsher, D.W. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 2018, 37, 5435–5450.
- Hammer, E.; Ernst, F.D.; Thiele, A.; Karanam, N.K.; Kujath, C.; Evert, M.; Völker, U.; Barthlen, W. Kidney protein profiling of Wilms’ tumor patients by analysis of formalin-fixed paraffin-embedded tissue samples. Clin. Chim. Acta 2014, 433, 235–241.
- Khaleel, S.; Katims, A.; Cumarasamy, S.; Rosenzweig, S.; Attalla, K.; Hakimi, A.A.; Mehrazin, R. Radiogenomics in clear cell renal cell carcinoma: A review of the current status and future directions. Cancers 2022, 14, 2085.
- Ferro, M.; Musi, G.; Marchioni, M.; Maggi, M.; Veccia, A.; Del Giudice, F.; Barone, B.; Crocetto, F.; Lasorsa, F.; Antonelli, A. Radiogenomics in Renal Cancer Management—Current Evidence and Future Prospects. Int. J. Mol. Sci. 2023, 24, 4615.
This entry is offline, you can click here to edit this entry!