Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Nipah virus is a negative-sense single-stranded ribonucleic acid ((−) ssRNA) virus within the family Paramyxoviridae. Nipah virus infection occurs predominantly in isolated regions of Malaysia, Bangladesh, and India in small outbreaks. Factors that affect animal–human disease transmission include viral mutation, direct contact, amplifying reservoirs, food, close contact, and host cell mutations. There are different strains of Nipah virus, and small outbreaks in humans limit known research and surveillance on this pathogen.
- Henipavirus
- Hendra
- innate
- adaptive
- immunology
- molecular
- Nipah
- Paramyxovirus
- virus
1. Introduction
Newly emerging viral infections remain a potential public health threat, including Henipaviruses discovered in the 1990s. Two of these are highly pathogenic in humans and are detailed below. During pathological research, Koch (1884) defined four key measures of microorganisms (microbes). Firstly, microbes should be found in abundance in disease and not in health; secondly, a microbe should be isolated from a host and grown in culture. In addition, a microbe should cause disease when infecting a host organism and be reisolated from inoculated or affected hosts to isolate the causal agent.
Within the binomial classification of kingdoms, Henipaviruses are defined under the kingdom Orthornavirae and order Mononegavirales, that include (−) ssRNA viruses within the family Paramyxoviridae (see Figure 1) [1][2].
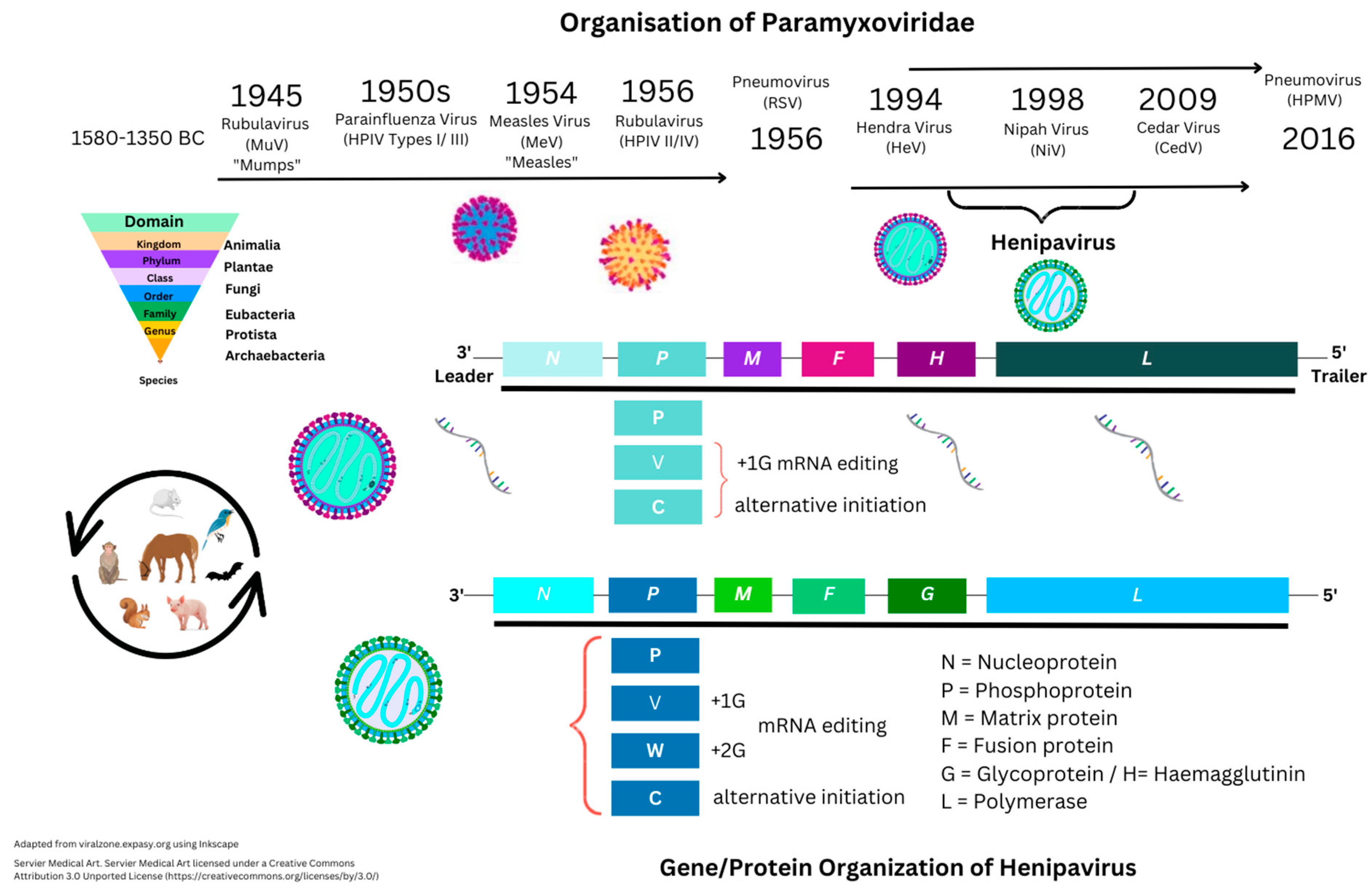
Figure 1. Historical structure perspectives of Paramyxoviridae.
In comparison, other viruses include (+) ssRNA viruses, with the differences being that (−) ssRNA viruses have complementary RNA and require transcription to produce (+) ssRNA to produce mRNA and then translation by RNA polymerase enzymes. Other categories include those of single- and double-stranded deoxyribonucleic viruses (ssDNA/dsDNA) [3][4]. Negative-sense ssRNA viruses historically include those that may cause unusual levels of disease or mortality in vertebrates. For example, Ebola virus, Hantavirus, Influenza, Lassa fever virus, and Rabies virus are only examined from tissue samples within Biosafety Level (BSL) 1–4 laboratories based on potential pathogen risk categorization (Figure 1). Nipah virus is categorized as a BSL4 pathogen.
The homologous Paramyxoviridae Hendra virus was first described in 1994, when initial reports appeared of a Morbillivirus in horses in Australia with high mortality of unknown origin [5]. In humans, these can cause encephalitis and/or respiratory disease. Nipah virus is named after the location of initial outbreaks in Malaysia around late 1998. Further outbreaks in humans and animals, and/or more specifically in livestock, have occurred in Australia, Malaysia, Singapore, Cambodia, and Bangladesh. Mortality has been seen with NiV infection in at least six species, including horses, cats, and dogs, as well as ferrets, hamsters, guinea pigs, and monkeys. Besides Malaysia/Singapore (1998–1999), other reports of NiV infection were documented in Bangladesh (2001/2003/2004). Similarly, outbreaks were also seen in India (2001/2007/2018), and more recently in 2021 [6]. Nipah virus is classified as a Biosafety Level 4 (BSL4) pathogen because of its high fatality rates, thought to be 35% or more, requiring further clarification. In the first outbreak (1998), within 265 individuals, it was indicated that fatality rates were around 40% initially. Data on Uniprot is suggestive that at least two of the following hosts have NiV infections: Cynopterus brachyotis (the lesser short-nosed fruit bat), Eonycteris spelaea (the lesser dawn bat), Macroglossus spelaeus (the cave nectar bat), Homo sapiens (the human), Pteropus vampyrus (the large flying fox bat), Scotophilus kuhlii (the lesser asiatic yellow bat), but was also dominant in Sus scrofa (the pig), with all viral genes seen in these species.
2. Epidemiological and Clinical Perspectives
2.1. Clinical Manifestations and Diagnosis
Nipah virus infection frequently causes severe disease and mortality, with symptoms that include encephalitis and excessively high infection fatality rates (IFR) like other BSL4 pathogens (e.g., Ebola (EBOV)). Sensory receptors for pathogens exist within all biological systems under investigation below with regards to NiV research. These receptors are expressed within the respiratory, nervous, circulatory, endocrine, reproductive, muscular, and skeletal systems. Researchers have examined systemic immune system cytokines, chemokines, and CD markers during SARS-CoV-2 infection in depth [7]. However, the sections below discuss the pathogenesis of NiV infection and immune system regulation.
Clinical symptoms may include headache, fever, and muscle aches, as well as fatigue. Muscle stiffness, confusion, agitation, seizure, speech, and hearing issues can occur. In children, nausea, irritability, poor feeding patterns, and rigor can be considered. Comparisons of NiV infections are made with Japanese encephalitis. Severe central nervous system (CNS) symptoms could be considered similar; however, this also includes the peripheral nervous system (PNS) and immune system regulation.
Early analysis indicated through brain magnetic resonance (MR) scans that the subcortical and deep white matter of the brain possessed bilateral foci. This was illustrated to potentially affect periventricular areas and the corpus callosum with critical lesions less than 1 cm in diameter that may differentiate NiV infection and Japanese encephalitis virus [8]. Other neurotropic viruses can be ruled out with cerebrospinal fluid (CSF) analysis indicative of lymphocyte infiltration (pleocytosis) [6]. Initial symptoms that can occur (4–14 days) are fever and headache, with concurrent signs of acquired respiratory distress syndrome (ARDS). Subsequent cough, sore throat, and difficulty breathing can also occur. Concerningly, compartmentalized brain tissue swelling (oedema) can occur from infection, trauma, or stroke. Encephalitis is considered inflammation derived from chronic viral or acute infection accompanied by drowsiness, disorientation, and mental confusion that can progress to a coma within 24–48 h [9]. Cell counts can clarify this further. Recent population studies clarify that this can vary according to age, country, season, viral genetic mutations, and immune status. At least two of the following symptoms were indicated in recent reports as being indicative of encephalitis: fever, seizures, or focal neurological findings. Concurrent brain parenchyma with CSF pleocytosis of more than four leukocytes per µL is suggested in recent reports [10][11][12]. Viral encephalitis can be characterised by a combination of systemic effects, including vasculitis and necrosis, in both the CNS and PNS [12]. Extensive cellular infection of neurons, endothelial cells, and smooth muscle cells of the vasculature can occur. Initial interstitial pneumonia, accompanied by syncytial cell formation, may be observed with mononuclear cellular infiltrates resembling other pathologies. Additionally, long-term sequelae in survivors may include encephalopathy, ocular motor palsy, cervical dystonia, and facial paralysis [13]. It is notable that the mean age of survivors was 14.5 years old in the prior report; this is therefore clearly indicative of severity in children with an infection similar to MeV. The Nipah virus can be described as a neurotropic virus. However, researchers examine the factors relevant to clarify and explain how T cell responses, a critical part of immune system development during childhood, can be considered further. Few reports describe the exact nature of adaptive T cell responses apart from those seen in Cytomegalovirus (CMV) and Respiratory Syncytial virus infections so far [14][15][16].
2.2. Human Transmission of Henipaviruses
Initial reports remain unclear about the routes of NiV transmission. This is likely to be a combination of oropharyngeal and nosocomial routes or through fomites. It was thought that NiV could be transmitted through the date palm until 2014, according to earlier outbreaks in Bangladesh [17]. Outbreaks in India (2001–2007) continued to illustrate the similar severity of NiV disease with high IFR occurrence (74–100%), but also recently in 2019 in Bangladesh with similar IFR [18]. Prior reports during the NiV outbreaks in Bangladesh were suggestive of the duration of illness leading to mortality at <6 days. Literature indicates that there are few reports of successful live NiV cultures apart from those obtained from samples of P. hypomelanus, P. vampyrus, and P. lylei bats. However, researchers in Australia (2011) investigated other Pteroptids, like P. alecto, and concurred that these could be considered natural host reservoirs where Henipaviruses co-evolve. It was indicated in samples analyzed for HeV or NiV infection that symptoms in bats appear to be subclinical with a low serological response [19]. Reverse transcriptase polymerase chain reaction (RT–PCR) is often utilised with cycle threshold (Ct) values compared for HeV genomes from urine, blood, throat, and rectal swab samples. The duration of PCR detection from urine HeV swabs is indicated at 10–19 days following infection in animals. In 2014, NiV was isolated from P. medius, with evidence that NiV spillover is indeed rare [20]. Importantly, during the initial Bangladesh (2004) outbreaks, NiV viral RNA was successfully isolated, meeting the above definition of a microbe [21].
2.3. Animal Host Reservoirs of Henipaviruses
Early outbreaks of NiV infection and concerns about pig vectors resulted in the slaughter of 1 million pigs due to viral severity. Recent reports have examined bat novel virus transmission in different species, indicating that species like Myotis spp. rather than Pteropid spp. could be reservoir hosts. Also, in an early surveillance study (n = 692), protein analysis indicated the NiV proteins below could be immunogenic. In a cross-sectional study of bat species, it was seen that bat samples (n = 33) possessed antibodies that were not cross-reactive against cellular infection with either HeV or NiV [22]. In a single-case isolation study, NiV RNA was detected in blood, bronchoalveolar lavage fluid (BALF), and CSF alongside serum IgM antibodies in people. In variable bat species, the immunoglobulin G (IgG) specific for NiV was indicated to be found in P. medius (21%), and R. leschenaultia (37.73%). Furthermore, neutralising antibodies (nAb) evidence of NiV infection were found in P. medius (20.68%) that were anti–NiV immunoglobulin G (IgG) antibodies. Thus far, the genomic clades NiV-M and NiV-B, which refer to the initial outbreaks in Malaysia and Bangladesh, have been identified. Sequencing recently was indicative of slight variations in genome size (15,100–18,172 bps) in Pteropus bats, with NiV not found in Rousettus spp. during surveillance [23]. Serology reports in bat reservoirs are variable. In a recent outbreak case study, surveillance of bats (2018/2019) confirmed that 3.2% and 25% of the samples obtained were seropositive for NiV infection. This indicated that bat immune responses, as indicated by serology, could plausibly be established at cyclical levels until 6–7 years after NiV infection [24]. Indeed, in animal surveillance prior to 2011, reports did indicate that bats may have differential regulation of immune responses, with further research required to investigate this [19].
3. Henipavirus Pathogenesis
3.1. Introduction to Nipah Virus Proteins and Genes
Currently, the Uniprot database shows 281 proteins listed under Henipavirus that include nine from the original HeV 1994 strain (AF017149) and eight from NiV strains (UP000128950). Nipah virus is known to be approximately 18,200 bps in size and encode six proteins. These are nucleocapsid (N), phosphoprotein (P), matrix protein (M), fusion protein (F), glycoprotein (G), and polymerase (L) protein. An additional NiV P protein encodes a further three non-structural proteins (NSP) responsible for RNA editing (V and W proteins) or with an open reading frame (ORF/C protein).
Recent outbreaks of NiV indicate that genome sequencing is required for the NiV infection that has occurred so far in isolated outbreak samples [23][24]. In 2019, in a case study, it was indicated that mutations occurred within the NiV N gene that the sequencing database sites (nextstrain.org) do not currently display; although massive global surveillance during the COVID–19 pandemic on viral mutations in SARS-CoV-2 spike (S) protein domains and other potential pathogens does continue [23][24].
3.2. Nipah Virus Viral Protein Factors
Two viral membrane glycoproteins above are considered to mediate NiV cellular attachment and include the haemagglutinin–neuraminidase (HN) proteins. These attach to the receptor binding protein (RBD) and are considered stalks shared amongst other Paramyxoviridae requiring fusion peptides (F) with NiV possessing a NiV G glycoprotein [25]. These two proteins are type II membrane glycoproteins composed of a cytoplasmic tail and transmembrane region attached to the viral envelope with a stalk and globular head. Insertion of the hydrophobic NiV F protein alongside the NiV G protein into the cellular plasma membrane is required for viral propagation. Nipah virus entry to host cells was described as utilising recombinant NiV N/NiV P interaction, that is crucial in disrupting NiV viral mRNA. This affects polymerase transcription and replicase activity through assembly and encapsulation into full-length genomic viral RNA [26][27]. Furthermore, the N-terminal protein of NiV N could be inhibited, as with SARS-CoV-2, where the conformation and binding shape of the unassembled NiV N protein affect cellular entry. Hendra virus protein homology gave early indications that IFN signaling inhibition occurred for both type I and type II IFNs (IFN–α/IFN–γ), that usually facilitates viral elimination. In 2003, the HeV V protein was thought to accumulate intracellularly with specific signal transducer and activator of transcription (STAT) proteins. These include STAT1 and others below that can be dysregulated, preventing further IFN-induced signaling [28]. Therefore, the NiV N protein, through saturation of a binding domain within the NiV P protein (polymerase complex), could affect enzyme activity through transcriptase or replicase activity [26][29]. Moreover, NiV M protein involvement in viral budding is considered to affect cellular morphology [30]. Nevertheless, F peptides are required to bind and traverse the phospholipid bilayer, dependently or independently of cellular receptors. Further clarification from Michael Weis’ group in 2015 (Marburg, Germany) indicated the NiV F protein contains Y525RSL (a tyrosine signal) that is essential for NiV F protein uptake, proteolytic activation, and endocytosis [31]. In comparison, Protein C researched in vivo can cause more serious respiratory damage during NiV infection as studied in knockout experiments [32]. In 2003, utilising an in vitro model of recombinant Newcastle Disease virus (NDV), all three NiV proteins (NiV V, NiV W, and NiV C) were indicated as required for intracellular NiV replication [33]. Cellular regulation of NiV appears to clarify in vitro that p38 is also essential [34][35]. Whilst X-ray crystallography illustrated that NiV M protein binds to intracellular phosphatidylserine (PS) and soluble cellular phosphatidylinositol 4,5-bisphosphate (PI (4,5) P2). These maintain and regulate intracellular NiV virion particle formation and egress through NiV M protein polymerization [36]. The larger NiV L protein function is that of a viral polymerase required for replication [37][38]. Viral NiV replication therefore occurs intracellularly through post-transcriptional modification, resulting in 5′ capping and methylation of RNA. Thereafter, NiV V protein is described as the virulence factor that may abrogate type I IFN responses intracellularly, as discussed below [39].
3.3. Receptor-Mediated Nipah Virus Entry
Initially, it was clarified that two ephrin receptor types, B2 and B3 (EFNB2/ENFB3), are both encoded by EFNB genes are implicated in cellular host HeV and NiV infection [40]. In 2003, EFNB2 receptor stability was clarified as being induced by the NiV G protein preceding full receptor activation, similar to a viroporin [41]. Ephrin B2 is preferentially expressed on vascular endothelial cells, as well as alveolar type II epithelial cells and around host naïve T cells (TN), with a known role in angiogenic tissue remodeling [42]. Moreover, according to current data, EFNB2 is constitutively expressed at higher levels in the brain, cerebral cortex, amygdala, basal ganglia, hypothalamus, and white matter. Ephrin B2 is conserved across species and could be a therapeutic target. It appears that two NiV proteins, NiV F and NiV G, co-stabilize at the EFNB2/EFNB3 receptor interface to facilitate viral RNA cellular entry [43]. Microbial and viral pathogens utilise protein glycosylation for host cell entry and immune response evasion. This can occur through either N-glycans or O-glycans affecting cellular protein binding and signaling at the cell membrane surface. Therefore, N-glycosylation in NiV infection was indicated by unique N-X-S/T motifs (asparagine (N), any amino acid apart from proline (X), serine (S), or threonine (T)). Currently, O-glycosylation of S/T residues remains unknown. Other Paramyxoviruses are affected by N-glycans in MeV infections that alter F protein-mediated fusion and processing. In addition, C–type lectins (e.g., galectin–1) bind to N and O glycans that inhibit infection but increase endothelial viral RNA entry [44][45]. This was investigated in 2015, when the disruption of eight predicted N-glycosylation sites was examined to find six (G2–G7), with one conserved between HeV and NiV in the stalk (G2) and five in the head domain (G3–G7) [46]. The EFNB2-predominant receptor is better characterised through histological analysis and is absent in other cellular subtypes, but other unknown receptors may exist [40][47]. As shown below, the NIV G protein appears to be stabilized as a trimer with NiV F/EFNB2 during cellular fusion. Intracellularly, NiV F is activated, endocytosed, and can be cleaved through proteases (e.g., cathepsin L), but remains active during viral replication and virion formation [48]. Given the discussion below and considering current immunogen development, it appears as though the NiV F protein may either be less immunogenic or shielded from immune cells during cellular infection.
3.4. Cellular Immunomodulatory Properties of Nipah Virus Infection
Bats have been described as having IFN modulatory properties that enable viral transmission. Reports appeared in 2012 describing bat viral reservoirs [49]. Other reviews document both OPXV infection and SARS-CoV-2 affecting host IFN cell synthesis. Comparatively, less is known about the role that IFN plays in the cellular regulation of viral infection. Factors that have been described in P. vampyrus include type I/III IFNs modulating IFN gene transcription through receptors; for example, different species may have different specific type I IFN subtypes (e.g., IFN–α/β) or type I IFN receptors (e.g., IFNAR–7). Other species like M. lucifugus and P. vampyrus may have more of the other type I IFN subtypes (IFN–ω). Earlier articles are indicative of a predominant type I/III IFN regulatory role in both human and bat species that can eliminate viral infections [49]. In comparison, type II IFN (IFN–γ) can also be considered important for adaptive immune responses in cancer [50][51]. Within NiV disease, there are suggestions that both NIV W and NIV V both appear to regulate IFN–β production [52]. This could occur by inhibiting the proteolysis of the UBXN1 protein and suppressing both retinoic acid-inducible gene I (RIG–I) and a similar helicase receptor (MDA–5). These stabilize mitochondrial antiviral signaling protein complexes (MAVS and ubiquitin-binding UBA domain/UBX motif, UBXN1) are known to regulate the nuclear transcription factor (NF–κB) [52][53]. Therefore, as NiV W can influence NF–κB, the detailed role of other pattern recognition receptors (PRR) like Toll-like receptors (TLRs) that are present on cell membranes and vesicles during NiV infection is starting to emerge. There are ten known TLRs in humans.
Interferon modulatory properties affect intracellular viral propagation and modulate immune responses that are well characterised in humans but less so in bats. These include interferon regulatory factors (IRF3/7) and TLRs (TLR3/4/7/8/9) known to be affected by viral DNA/RNA host cell entry. Many other PRRs are considered better known in other pathologies [54]. Early in NiV research, NiV V and NiV W proteins were seen in vitro to selectively antagonize host cell IFN gene transcripts. In the context of NiV infection, TLR 7/9 were plausibly seen to be inhibited, thereby additionally reducing cellular IFN–α synthesis and induction through the IκB kinase (IKK–α) and IRF7 pathways by inhibiting NF–κB transcription [32][55]. Very recently, MAVS were thought to be critical alongside both TLR7/MyD88, that could be redundant during NiV infection rather than TLR3. These recent reports indicate the biological plausibility that TLR7 on immune cells could affect activated T cell subtypes known to be preferentially infected and that express high levels of EFNB2 during active NiV infection [56]. The amino terminals of NiV V and NiV W proteins may be causal in this, as the NiV V protein carboxyl terminus is known to be relevant to HPIV-II infection [33]. Recently, mRNA transcripts were noted during in vivo NiV immunogen experiments. These researchers noted upregulation of transcription factors (TFs) relevant to IFN responses (IFIT2, GBP1, IFIT1, MX1, OASL, and IFIT3). However, other immune response genes were also noted as upregulated (e.g., FCAR, CLEC4E, and IL-8) [57]. However, as NiV V and NiV W diffuse intracellularly to the nucleus, these may inhibit the janus kinase (JAK) and STAT protein pathways affecting the IFN–β promoter and IRF3. Furthermore, extracellular NiV W in return may antagonize TLR3 around plasma cell membranes and intracellular vesicles. Below are shown known cellular interactions during NiV infection and upregulated molecular gene transcripts observed in research so far in italics (see Figure 2).
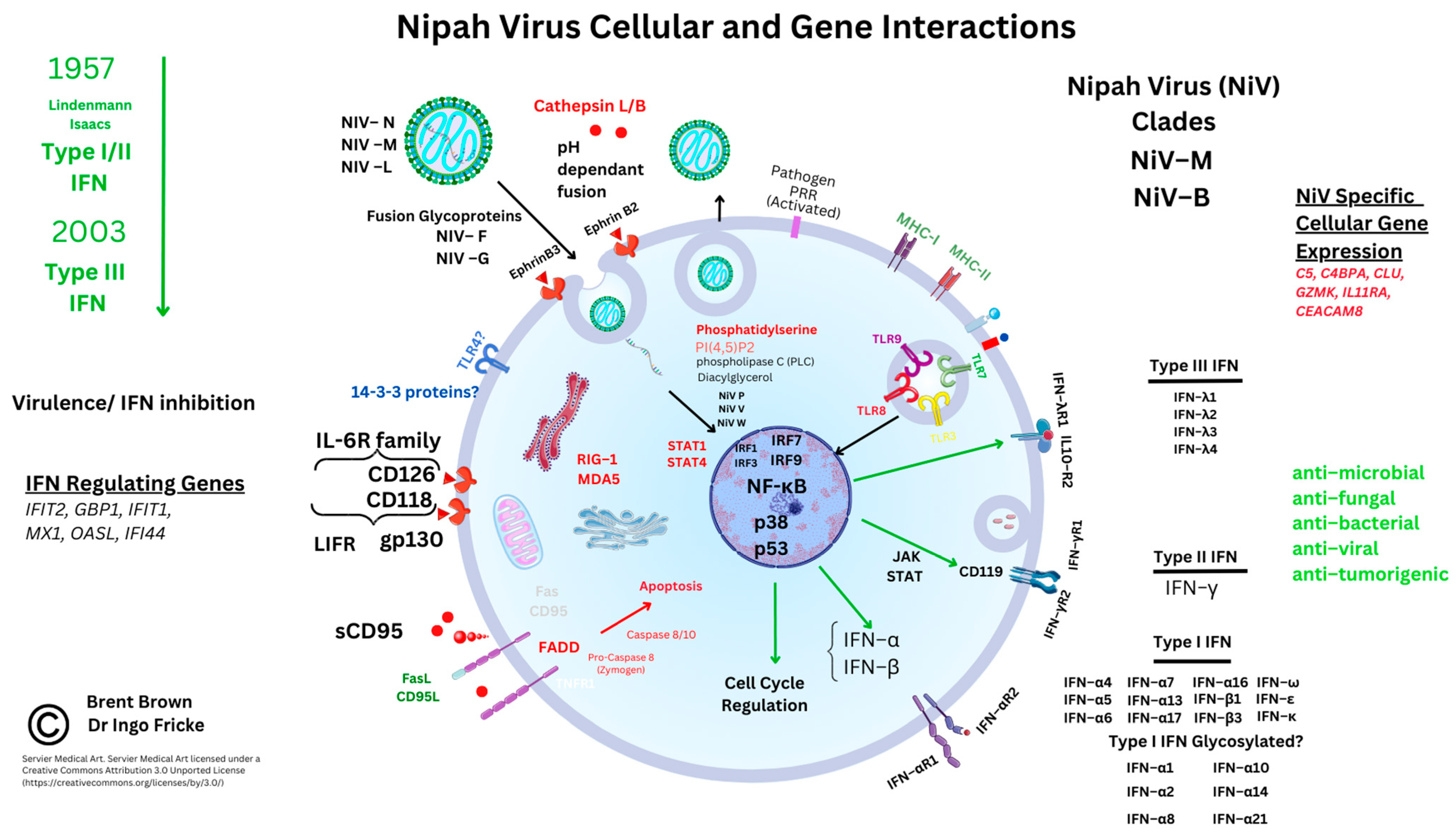
Figure 2. Nipah virus molecular and cellular regulation.
Other RNA viruses can affect TLR3 through Toll-interleukin–1 receptor (TIR) domain-containing adaptor-inducing IFN–β (TRIFs) [58][59]. Recent reports are indicative that IRF1 also plays a role in IFN regulation in both monocytes and macrophages (Mϕ), together with IRF7/9 coordinating TLR sensing of viral DNA/RNA [60][61]. Furthermore, IFN regulation appears to be regulated through two NiV proteins, NiV V and NiV M, that enhance the tripartite motif (TRIM6) post-translational protein degradation. This could impair the oligomerization and phosphorylation of an IκB kinase (IKK–ε) [62]. As noted above, IFN–β is required for myeloid cell differentiation and maturation of antigen-presenting cells (dendritic cells (DCs), monocytes, and Mϕ), as well as regulating Mϕ apoptosis [63][64]. Data indicates that TRIM6 is also constitutively expressed predominantly by neutrophils within the immune system. Furthermore, STAT proteins were examined through immunoprecipitation of haemagglutinin (HA) proteins tagged to NiV P, NiV V, and NiV W proteins. These were flanked by a peptide sequence (FLAG, denoted by the DYKDDDDK sequence). The NiV P protein was observed to have a conserved interaction with STAT1, STAT2, and STAT4, but specifically with amino acid residues (114–140) of the NiV P protein. Whilst STAT1/4, but more specifically the STAT1 SH2 domain can affect IFN–β nuclear transcription. In addition, it was found that NiV P/V/W proteins could also antagonize STAT4 through N-terminal domains [65]. Moreover, within endothelial cells, STAT4 protein in vitro research indicates that IFN-α rather than the DC maturation cytokine IL-12 is required to activate STAT4 alongside the CD4 TH1 cell response [66].
This entry is adapted from the peer-reviewed paper 10.3390/immuno3020011
References
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the Order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980.
- Latorre, V.; Mattenberger, F.; Geller, R. Chaperoning the Mononegavirales: Current Knowledge and Future Directions. Viruses 2018, 10, 699.
- Chen, Y.G.; Hur, S. Cellular Origins of DsRNA, Their Recognition and Consequences. Nat. Rev. Mol. Cell Biol. 2022, 23, 286–301.
- Van Etten, J.L.; Lane, L.C.; Dunigan, D.D. DNA Viruses: The Really Big Ones (Viruses). Annu. Rev. Microbiol. 2010, 64, 83–99.
- Murray, K.; Rogers, R.; Selvey, L.; Selleck, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Hooper, P.; Westbury, H. A Novel Morbillivirus Pneumonia of Horses and Its Transmission to Humans. Emerg. Infect. Dis. 1995, 1, 31–33.
- Farrar, J.J. Nipah-Virus Encephalitis—Investigation of a New Infection. Lancet 1999, 354, 1222–1223.
- Brown, B.; Ojha, V.; Fricke, I.; Al-Sheboul, S.A.; Imarogbe, C.; Gravier, T.; Green, M.; Peterson, L.; Koutsaroff, I.P.; Demir, A.; et al. Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms. Vaccines 2023, 11, 408.
- Chadha, M.S.; Comer, J.A.; Lowe, L.; Rota, P.A.; Rollin, P.E.; Bellini, W.J.; Ksiazek, T.G.; Mishra, A.C. Nipah Virus-Associated Encephalitis Outbreak, Siliguri, India. Emerg. Infect. Dis. 2006, 12, 235–240.
- Fisher, D.L.; Defres, S.; Solomon, T. Measles-Induced Encephalitis. QJM 2015, 108, 177–182.
- Lee, S.J.; Kim, J.M.; Keum, H.R.; Kim, S.W.; Baek, H.S.; Byun, J.C.; Kim, Y.K.; Kim, S.; Lee, J.M. Seasonal Trends in the Prevalence, and Incidence of Viral Encephalitis in Korea (2015–2019). J. Clin. Med. 2023, 12, 2003.
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A Clinical Approach to Diagnosis of Autoimmune Encephalitis. Lancet Neurol. 2016, 15, 391–404.
- Tunkel, A.R.; Glaser, C.A.; Bloch, K.C.; Sejvar, J.J.; Marra, C.M.; Roos, K.L.; Hartman, B.J.; Kaplan, S.L.; Scheld, W.M.; Whitley, R.J. The Management of Encephalitis: Clinical Practice Guidelines by the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 47, 303–327.
- Sejvar, J.J.; Hossain, J.; Saha, S.K.; Gurley, E.S.; Banu, S.; Hamadani, J.D.; Faiz, M.A.; Siddiqui, F.M.; Mohammad, Q.D.; Mollah, A.H.; et al. Long-Term Neurological and Functional Outcome in Nipah Virus Infection. Ann. Neurol. 2007, 62, 235–242.
- Roumanes, D.; Falsey, A.R.; Quataert, S.; Secor-Socha, S.; Lee, F.E.-H.; Yang, H.; Bandyopadhyay, S.; Holden-Wiltse, J.; Topham, D.J.; Walsh, E.E. T-Cell Responses in Adults During Natural Respiratory Syncytial Virus Infection. J. Infect. Dis. 2018, 218, 418–428.
- Fülöp, T.; Larbi, A.; Pawelec, G. Human T Cell Aging and the Impact of Persistent Viral Infections. Front. Immunol. 2013, 4, 271.
- Hassoun, F.; Goldeck, D.; Pera, A.; van Heemst, D.; Slagboom, P.E.; Pawelec, G.; Solana, R. Functional Changes of T-Cell Subsets with Age and CMV Infection. Int. J. Mol. Sci. 2021, 22, 9973.
- Islam, M.S.; Sazzad, H.M.S.; Satter, S.M.; Sultana, S.; Hossain, M.J.; Hasan, M.; Rahman, M.; Campbell, S.; Cannon, D.L.; Ströher, U.; et al. Nipah Virus Transmission from Bats to Humans Associated with Drinking Traditional Liquor Made from Date Palm Sap, Bangladesh, 2011–2014. Emerg. Infect. Dis. 2016, 22, 664–670.
- Arankalle, V.A.; Bandyopadhyay, B.T.; Ramdasi, A.Y.; Jadi, R.; Patil, D.R.; Rahman, M.; Majumdar, M.; Banerjee, P.S.; Hati, A.K.; Goswami, R.P.; et al. Genomic Characterization of Nipah Virus, West Bengal, India. Emerg. Infect. Dis. 2011, 17, 907–909.
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Henipavirus Ecology Research Group. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951.
- Anderson, D.E.; Islam, A.; Crameri, G.; Todd, S.; Islam, A.; Khan, S.U.; Foord, A.; Rahman, M.Z.; Mendenhall, I.H.; Luby, S.P.; et al. Isolation and Full-Genome Characterization of Nipah Viruses from Bats, Bangladesh. Emerg. Infect. Dis. 2019, 25, 166–170.
- Hassan, M.Z.; Sazzad, H.M.S.; Luby, S.P.; Sturm-Ramirez, K.; Bhuiyan, M.U.; Rahman, M.Z.; Islam, M.M.; Ströher, U.; Sultana, S.; Kafi, M.A.H.; et al. Nipah Virus Contamination of Hospital Surfaces during Outbreaks, Bangladesh, 2013–2014. Emerg. Infect. Dis. 2018, 24, 15–21.
- Li, Y.; Wang, J.; Hickey, A.C.; Zhang, Y.; Li, Y.; Wu, Y.; Zhang, H.; Yuan, J.; Han, Z.; McEachern, J.; et al. Antibodies to Nipah or Nipah-like Viruses in Bats, China. Emerg. Infect. Dis. 2008, 14, 1974–1976.
- Sudeep, A.B.; Yadav, P.D.; Gokhale, M.D.; Balasubramanian, R.; Gupta, N.; Shete, A.; Jain, R.; Patil, S.; Sahay, R.R.; Nyayanit, D.A.; et al. Detection of Nipah Virus in Pteropus Medius in 2019 Outbreak from Ernakulam District, Kerala, India. BMC Infect. Dis. 2021, 21, 162.
- Yadav, P.D.; Sahay, R.R.; Balakrishnan, A.; Mohandas, S.; Radhakrishnan, C.; Gokhale, M.D.; Balasubramanian, R.; Abraham, P.; Gupta, N.; Sugunan, A.P.; et al. Nipah Virus Outbreak in Kerala State, India Amidst of COVID-19 Pandemic. Front. Public Health 2022, 10, 14.
- Adu-Gyamfi, E.; Kim, L.S.; Jardetzky, T.S.; Lamb, R.A. Mutagenesis of Paramyxovirus Hemagglutinin-Neuraminidase Membrane-Proximal Stalk Region Influences Stability, Receptor Binding, and Neuraminidase Activity. J. Virol. 2016, 90, 7778–7788.
- Ranadheera, C.; Proulx, R.; Chaiyakul, M.; Jones, S.; Grolla, A.; Leung, A.; Rutherford, J.; Kobasa, D.; Carpenter, M.; Czub, M. The Interaction between the Nipah Virus Nucleocapsid Protein and Phosphoprotein Regulates Virus Replication. Sci. Rep. 2018, 8, 15994.
- Bossart, K.N.; Fusco, D.L.; Broder, C.C. Paramyxovirus entry. Adv. Exp. Med. Biol. 2013, 790, 95–127.
- Rodriguez, J.J.; Wang, L.-F.; Horvath, C.M. Hendra Virus V Protein Inhibits Interferon Signaling by Preventing STAT1 and STAT2 Nuclear Accumulation. J. Virol. 2003, 77, 11842–11845.
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.-M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.H.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah Virus Unassembled Nucleoprotein in Complex with Its Viral Chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759.
- Liu, Q.T.; Wang, Q.; Zhang, Y.; Kliemke, V.; Liu, Q.; Chou, K.C. The Nanoscale Organization of Nipah Virus Matrix Protein Revealed by Super-Resolution Microscopy. Biophys. J. 2022, 121, 2290–2296.
- Weis, M.; Maisner, A. Nipah Virus Fusion Protein: Importance of the Cytoplasmic Tail for Endosomal Trafficking and Bioactivity. Eur. J. Cell Biol. 2015, 94, 316–322.
- Satterfield, B.A.; Cross, R.W.; Fenton, K.A.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Graber, J.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. Nipah Virus C and W Proteins Contribute to Respiratory Disease in Ferrets. J. Virol. 2016, 90, 6326–6343.
- Park, M.-S.; Shaw, M.L.; Muñoz-Jordan, J.; Cros, J.F.; Nakaya, T.; Bouvier, N.; Palese, P.; García-Sastre, A.; Basler, C.F. Newcastle Disease Virus (NDV)-Based Assay Demonstrates Interferon-Antagonist Activity for the NDV V Protein and the Nipah Virus V, W, and C Proteins. J. Virol. 2003, 77, 1501–1511.
- Stachowiak, B.; Weingartl, H.M. Nipah Virus Infects Specific Subsets of Porcine Peripheral Blood Mononuclear Cells. PLoS ONE 2012, 7, e30855.
- Wang, L.; Xia, Z.; Tang, W.; Sun, Y.; Wu, Y.; Kwok, H.F.; Sun, F.; Cao, Z. P38 Activation and Viral Infection. Expert Rev. Mol. Med. 2022, 24, e4.
- Norris, M.J.; Husby, M.L.; Kiosses, W.B.; Yin, J.; Saxena, R.; Rennick, L.J.; Heiner, A.; Harkins, S.S.; Pokhrel, R.; Schendel, S.L.; et al. Measles and Nipah Virus Assembly: Specific Lipid Binding Drives Matrix Polymerization. Sci. Adv. 2022, 8, eabn1440.
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.-F. Hendra and Nipah Viruses: Different and Dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35.
- Talukdar, P.; Dutta, D.; Ghosh, E.; Bose, I.; Bhattacharjee, S. Molecular Pathogenesis of Nipah Virus. Appl. Biochem. Biotechnol. 2023, 195, 2451–2462.
- Tham, M.L.; Yusoff, K.; Othman, S.; Chia, S.L. V Protein, the Virulence Factor across the Family Paramyxoviridae: A Review. Asia Pac. J. Mol. Biol. Biotechnol. 2019, 27, 73–85.
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.-F.; Eaton, B.T.; et al. Ephrin-B2 Ligand Is a Functional Receptor for Hendra Virus and Nipah Virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657.
- Wong, J.J.W.; Young, T.A.; Zhang, J.; Liu, S.; Leser, G.P.; Komives, E.A.; Lamb, R.A.; Zhou, Z.H.; Salafsky, J.; Jardetzky, T.S. Monomeric EphrinB2 Binding Induces Allosteric Changes in Nipah Virus G That Precede Its Full Activation. Nat. Commun. 2017, 8, 781.
- Bochenek, M.L.; Dickinson, S.; Astin, J.W.; Adams, R.H.; Nobes, C.D. Ephrin-B2 Regulates Endothelial Cell Morphology and Motility Independently of Eph-Receptor Binding. J. Cell Sci. 2010, 123, 1235–1246.
- Sawatsky, B.; Grolla, A.; Kuzenko, N.; Weingartl, H.; Czub, M. Inhibition of Henipavirus Infection by Nipah Virus Attachment Glycoprotein Occurs without Cell-Surface Downregulation of Ephrin-B2 or Ephrin-B3. J. Gen. Virol. 2007, 88, 582–591.
- Bradel-Tretheway, B.G.; Liu, Q.; Stone, J.A.; McInally, S.; Aguilar, H.C. Novel Functions of Hendra Virus G N-Glycans and Comparisons to Nipah Virus. J. Virol. 2015, 89, 7235–7247.
- Stone, J.A.; Nicola, A.V.; Baum, L.G.; Aguilar, H.C. Multiple Novel Functions of Henipavirus O-Glycans: The First O-Glycan Functions Identified in the Paramyxovirus Family. PLoS Pathog. 2016, 12, e1005445.
- Bender, R.R.; Muth, A.; Schneider, I.C.; Friedel, T.; Hartmann, J.; Plückthun, A.; Maisner, A.; Buchholz, C.J. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 2016, 12, e1005641.
- Negrete, O.A.; Wolf, M.C.; Aguilar, H.C.; Enterlein, S.; Wang, W.; Mühlberger, E.; Su, S.V.; Bertolotti-Ciarlet, A.; Flick, R.; Lee, B. Two Key Residues in EphrinB3 Are Critical for Its Use as an Alternative Receptor for Nipah Virus. PLoS Pathog. 2006, 2, e7.
- Navaratnarajah, C.K.; Generous, A.R.; Yousaf, I.; Cattaneo, R. Receptor-Mediated Cell Entry of Paramyxoviruses: Mechanisms, and Consequences for Tropism and Pathogenesis. J. Biol. Chem. 2020, 295, 2771–2786.
- Baker, M.L.; Schountz, T.; Wang, L.-F. Antiviral Immune Responses of Bats: A Review. Zoonoses Public Health 2013, 60, 104–116.
- Ivashkiv, L.B. IFNγ: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558.
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101.
- Uchida, S.; Horie, R.; Sato, H.; Kai, C.; Yoneda, M. Possible Role of the Nipah Virus V Protein in the Regulation of the Interferon Beta Induction by Interacting with UBX Domain-Containing Protein1. Sci. Rep. 2018, 8, 7682.
- Hu, Y.; O’Boyle, K.; Auer, J.; Raju, S.; You, F.; Wang, P.; Fikrig, E.; Sutton, R.E. Multiple UBXN Family Members Inhibit Retrovirus and Lentivirus Production and Canonical NFκΒ Signaling by Stabilizing IκBα. PLoS Pathog. 2017, 13, e1006187.
- Banerjee, A.; Baker, M.L.; Kulcsar, K.; Misra, V.; Plowright, R.; Mossman, K. Novel Insights Into Immune Systems of Bats. Front. Immunol. 2020, 11, 26.
- Pisanelli, G.; Pagnini, U.; Iovane, G.; García-Sastre, A. Type I and Type II Interferon Antagonism Strategies Used by Paramyxoviridae: Previous and New Discoveries, in Comparison. Viruses 2022, 14, 1107.
- Iampietro, M.; Aurine, N.; Dhondt, K.P.; Dumont, C.; Pelissier, R.; Spanier, J.; Vallve, A.; Raoul, H.; Kalinke, U.; Horvat, B. Control of Nipah Virus Infection in Mice by the Host Adaptors Mitochondrial Antiviral Signaling Protein (MAVS) and Myeloid Differentiation Primary Response 88 (MyD88). J. Infect. Dis. 2020, 221, S401–S406.
- Foster, S.L.; Woolsey, C.; Borisevich, V.; Agans, K.N.; Prasad, A.N.; Deer, D.J.; Geisbert, J.B.; Dobias, N.S.; Fenton, K.A.; Cross, R.W.; et al. A recombinant VSV-vectored vaccine rapidly protects nonhuman primates against lethal Nipah virus. disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2200065119.
- Yoneda, M.; Guillaume, V.; Sato, H.; Fujita, K.; Georges-Courbot, M.-C.; Ikeda, F.; Omi, M.; Muto-Terao, Y.; Wild, T.F.; Kai, C. The Nonstructural Proteins of Nipah Virus Play a Key Role in Pathogenicity in Experimentally Infected Animals. PLoS ONE 2010, 5, e12709.
- Chen, Y.; Lin, J.; Zhao, Y.; Ma, X.; Yi, H. Toll-like Receptor 3 (TLR3) Regulation Mechanisms and Roles in Antiviral Innate Immune Responses. J. Zhejiang Univ.-Sci. B 2021, 22, 609–632.
- Irving, A.T.; Zhang, Q.; Kong, P.-S.; Luko, K.; Rozario, P.; Wen, M.; Zhu, F.; Zhou, P.; Ng, J.H.J.; Sobota, R.M.; et al. Interferon Regulatory Factors IRF1 and IRF7 Directly Regulate Gene Expression in Bats in Response to Viral Infection. Cell Rep. 2020, 33, 108345.
- Song, R.; Gao, Y.; Dozmorov, I.; Malladi, V.; Saha, I.; McDaniel, M.M.; Parameswaran, S.; Liang, C.; Arana, C.; Zhang, B.; et al. IRF1 Governs the Differential Interferon-Stimulated Gene Responses in Human Monocytes and Macrophages by Regulating Chromatin Accessibility. Cell Rep. 2021, 34, 108891.
- Bharaj, P.; Wang, Y.E.; Dawes, B.E.; Yun, T.E.; Park, A.; Yen, B.; Basler, C.F.; Freiberg, A.N.; Lee, B.; Rajsbaum, R. The Matrix Protein of Nipah Virus Targets the E3-Ubiquitin Ligase TRIM6 to Inhibit the IKKε Kinase-Mediated Type-I IFN Antiviral Response. PLoS Pathog. 2016, 12, e1005880.
- Kumaran Satyanarayanan, S.; El Kebir, D.; Soboh, S.; Butenko, S.; Sekheri, M.; Saadi, J.; Peled, N.; Assi, S.; Othman, A.; Schif-Zuck, S.; et al. IFN-β Is a Macrophage-Derived Effector Cytokine Facilitating the Resolution of Bacterial Inflammation. Nat. Commun. 2019, 10, 3471.
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin. Cancer Res. 2011, 17, 2619–2627.
- Keiffer, T.R.; Ciancanelli, M.J.; Edwards, M.R.; Basler, C.F. Interactions of the Nipah Virus P, V, and W Proteins across the STAT Family of Transcription Factors. mSphere 2020, 5, e00449-20.
- Torpey, N.; Maher, S.E.; Bothwell, A.L.M.; Pober, J.S. Interferon α but Not Interleukin 12 Activates STAT4 Signaling in Human Vascular Endothelial Cells. J. Biol. Chem. 2004, 279, 26789–26796.
This entry is offline, you can click here to edit this entry!