1. Introduction
The history of Q fever, the disease caused by
Coxiella burnetii, can be traced back to 1937, when it was described by Edward Holbrooke Derrick in Australia [
1]. Almost simultaneously, in the United States, an unknown agent isolated from ticks recovered from Nine Mile Creak region, Montana, was described [
2]. Australian and American teams shared their findings and concluded that they were studying the same agent and the same disease [
3]. The potential risk of Q fever to public health and the large gaps in the knowledge of this disease were recognized early, namely by the World Health Organization (WHO) that, in 1950, encouraged the epidemiological research. Consequently, Q fever was reported in 51 countries from the five continents [
4]. In Europe, Q fever was first reported in Greece, during the Second World War, in German soldiers who had febrile illness, the so-called “Balkan flu” [
5].
Nowadays, except in New Zealand,
C. burnetii is found worldwide, infecting a wide range of domestic and wildlife animals [
6,
7]. Q fever is listed in the Terrestrial Animal Health Code of the World Organization for Animal Health (WOAH) and all Member Countries are required to report the occurrence of the disease [
8].
Since its first report, human Q fever outbreaks have been regularly reported throughout the world [
9]. From 2007 until 2010, The Netherlands faced the largest Q fever outbreak ever recorded, resulting in over 4000 reported and 40,000 estimated infected people [
10]. This occurrence alerted public health authorities regarding
C. burnetii and the need for a harmonized monitoring of infection was highlighted [
11,
12,
13,
14]. In fact, during the last decade, the number of relevant publications on this subject increased significantly [
15].
Despite the wide host range of
C. burnetii, the infection is mostly recognized in domestic ruminants [
7,
16,
17,
18,
19]. However, over time, human Q fever outbreaks have often been related to spill-over infection from goats to humans, as shown in
Table 1.
2. Coxiella burnetii: The Microorganism and Its Pathogenesis
When Q fever was first described, its causative agent was unknown. In 1948, the genus
Coxiella was created and
Coxiella burnetii (Philip, 1948) was listed in the 6th edition of Bergey’s Manual of Determinative Bacteriology [
3,
33] as the aetiological agent of Q fever.
Phylogenetic investigations based on 16S rRNA sequence analysis placed
C. burnetii in the gamma group of proteobacteria, belonging to the order Legionellales, family Coxiellaceae, and genus
Coxiella [
34]. The first complete genome sequence of
C. burnetii was published in 2003. It corresponded to the original strain (RSA 493 strain) firstly isolated from ticks in the United States, also known as the Nine Mile strain. This event led to significant advances in the knowledge of
C. burnetii [
35]. The genome of
C. burnetii contains conserved genomic regions as well as polymorphic regions [
36]. Furthermore, the insertion sequence IS1111 plays an important role in the genomic plasticity of
C. burnetii. The number of IS1111 elements is highly variable between strains; many different genetic locations are described, showing a direct impact on
C. burnetii genotypes [
37].
C. burnetii is a small pleomorphic Gram-negative rod, presenting 0.2–0.4 μm wide and 0.4–1.0 μm long [
38]. All the lipopolysaccharides (LPSs) encoding genes are in a 38 Kb region in the
C. burnetii genome, and it has been observed that mutational variations in this region result in antigenic and virulence shift, termed “phase variation”. Antigenic variation results from an irreversible modification from smooth-type (phase I) to rough-type (phase II) LPS causing a dramatic reduction in virulence [
39]. Thus, the avirulent rough LPS (phase II) results from a point/frameshift mutation, small deletion, or transposon insertion in a gene in the LPS biosynthetic pathway [
40,
41]. Therefore, the sugar composition of phase II LPS is quite different because sugars such as L-virenose dihydrohydroxystreptose and galactosamine uronyl-(1,6) glucosamine are lacking [
39,
42]. So, the lack of virulence is associated with a shorter LPS and not with a defect in the synthesis of other virulence factors. However, it is interesting to note that avirulent forms of other strains besides Nine Mile show different patterns of deletions/mutations, suggesting that the biosynthesis of LPS in
C. burnetii is not yet completely understood [
40]. The shift from virulent phase I to avirulent phase II is likely due to repeated passages of the strains in cell cultures or embryonated eggs [
43].
Phase I
C. burnetii can be recovered from infected hosts and the smooth-type LPS of phase I disturbs an effective immune response, giving the phase I bacterium the opportunity to survive and multiply in the host cells. Therefore, phase I
C. burnetii is highly infectious [
39].
C. burnetii exhibits a biphasic developmental cycle in which two main morphological forms are identified: large cell variant (LCV) and small cell variant (SCV) [
44]. LCVs have a larger size (>0.5 μm), they are metabolically active, and have less electron dense forms. They have dispersed and filamentous chromatin and possess clearly distinguishable outer and cytoplasmic membranes with exposed LPS on the surface, sharing features with Gram-negative bacteria. These LCVs are sensitive to the decrease in osmotic pressure [
45,
46,
47]. SCVs are small rod-shaped forms ranging typically from 0.2 and 0.5 μm, being filterable through 0.22 μm filters. They are very compact and present low metabolic activity [
44,
46]. Some structural characteristics of SCVs are the electron-dense and condensed chromatin and the unusual cell envelope characterized by a high number of cross-links in peptidoglycans, which seems to enhance environmental stability [
45,
48]. Thus, they are very stable in the environment, showing a high resistance to osmotic, mechanical, chemical, heat, and desiccation stresses [
44,
48].
The primary target cells of
C. burnetii are blood-circulating monocytes, macrophages (e.g., lymph nodes, spleen, liver, and lungs) [
49], and trophoblasts in pregnant females [
50].
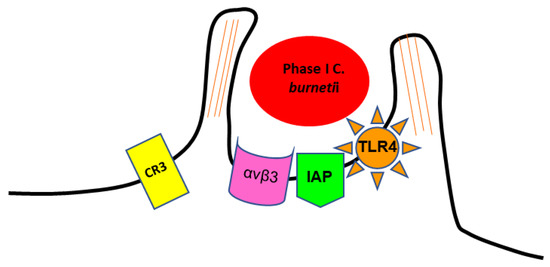
The internalisation of phase I SCV of
C. burnetii in target cells involves the recognition of several receptors [
51]. It is mediated by the leukocyte response integrin (LRI) (αvβ3) and an integrin-associated protein (IAP) [
39,
52]. The entry occurs through a microfilament-dependent endocytosis [
44,
51]. Phase I LPS induces a rearrangement of F-actin cytoskeleton, leading to pronounced membrane protrusions at the site of bacterial adherence. This phenomenon, called membrane ruffling, requires contact between
C. burnetii and host cells, and depends on the expression of toll-like receptor type 4 (TLR4) on the host cell surface (
Figure 1) [
53,
54,
55]. The ability to use αvβ3 integrin for invasion might be exploited by
C. burnetii as a mechanism to avoid the induction of an inflammatory response, as αvβ3 integrin is typically involved in the removal of apoptotic cells via phagocytosis, being generally associated with an inhibition of inflammation [
52]. Thus,
C. burnetii enters the cells without alerting the immune system [
56].
Figure 1. Scheme representing the internalization of phase I SCV of C. burnetii by monocyte-like cells.
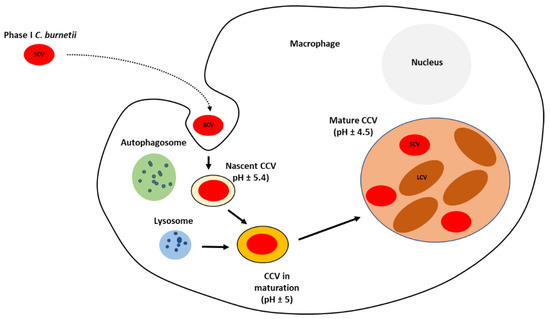
After internalization, bacteria localize within the nascent
Coxiella-containing vacuole (CCV), which traffics through the endocytic cascade. It develops into an early phagosome acquiring the small GTPase RAB5. This GTPase stimulates the fusion with early endosomes, resulting in acidification of the lumen to approximately pH 5.4 and acquisition of the early-endosomal marker protein 1 (EEA1) [
57,
58]. Early phagosome is converted into late phagosome acquiring acid hydrolases, which are involved in pronounced degradative activity, which
C. burnetii can resist [
59]. This late phagosome lacks RAB5 and EEA1 but acquires lysosome-associated membrane protein 1, 2, and 3 (LAMP1, LAMP2, and LAMP3) and vacuolar ATPase, which pumps protons into the maturing phagosome to further decrease the luminal pH to about 5.0 [
58,
60,
61].
C. burnetii persists and replicates, at a slow rate, within the large CCV with an acidic environment [
39,
62,
63]. The process of phagosome maturation continues with its fusion with lysosomal compartments to acquire cathepsins and hydrolases. The vacuolar ATPase further reduces the pH to around 4.5 [
58,
64]. Phagosome maturation depends on the balance between pro-inflammatory (IFN-γ, IL-12, and IL-6) and anti-inflammatory (IL-10) cytokines [
65].
C. burnetii modulates the genesis of CCV and has several strategies for adaptation to the stressful environment. It encodes a significant number of basic proteins that are probably involved in buffering the acidic environment of the CCV. Moreover, four sodium–proton exchangers and transporters for osmoprotectants are codified in its genome, allowing this bacterium to confront osmotic and oxidative stresses [
35].
During its biogenesis process, CCV becomes large and contains a large number of bacteria [
62].
C. burnetii does not synthesize its own CCV membrane. Multiple fusion events with autophagosomes along with endolysosomal vacuoles are essential to provide sufficient membrane to enlarge the CCV [
66,
67].
C. burnetii continuously directs fusion with other host cell compartments and inhibits apoptotic cell death, allowing a prolonged infectious cycle [
63,
68,
69,
70,
71].
The internalised SCV, within the CCV, suffers a differentiation into replicative and metabolically active LCV (
Figure 2). The low intra-phagosomal pH and perhaps enzyme system and/or nutrient sources present in the vacuole seem to trigger this differentiation. Lag phase extends to approximately two days post-infection and is composed primarily of SCV to LCV morphogenesis. The exponential phase occurs over the next four days with CCV harbouring replicating LCV almost exclusively. The LCV multiplies and persists within an expanding CCV that contains lysosomal elements, including an acid pH (5.0) and degradative proteases [
44,
46,
59,
72].
Figure 2. Diagram of the intracellular lifecycle of C. burnetii. CCV—Coxiella-containing vacuole; SCV—small cell variant; LCV—large cell variant.
A dramatic expansion of the CCV occurs concomitantly with the appearance of replicating LCV, occupying nearly the entire cytoplasm [
44,
66]. These metabolically active LCVs also play an important role in cell-to-cell spread during acute infection. This process is facilitated by the display of unique LCV antigens such as a porin protein termed P1. The stationary phase begins six days post-infection, concomitantly with the re-appearance of SCV. Following the accumulation of large numbers of LCVs,
C. burnetii converts back into SCVs, which are released from heavily infected cells by an undefined mechanism [
44].
The resistance properties of these SCVs strongly implicate this form as responsible for long-term extracellular survival and aerosol transmission of
C. burnetii [
44,
45].
3. Infection and Clinical Outcomes in Goats
It is globally recognized that
C. burnetii infection occurs mainly by inhalation of contaminated aerosols and, because
C. burnetii is a highly infective pathogen, low doses cause a high risk of illness [
73,
74]. So far, experimental studies on goats were not focused on estimating the infectious dose. However, in humans, it was estimated that the 50% infectious dose was around one bacterium [
75].
Alveolar macrophages are the first-line defence that confronts
C. burnetii [
49,
76]. The ability of these cells to rapidly respond recruiting additional immune cells is central for an effective antibacterial response in early stages of infection [
65,
77]. In primary infections, after entry into the organism, a bacteraemia occurs, leading to a systemic infection with the involvement of organs such as liver, spleen, lungs, and bone marrow [
38]. The organism can subsequently disseminate to colonize and replicate in resident macrophages of different tissues and organs [
78]. In pregnant goats, the main target cells are the trophoblasts in the allanthocorion, causing a placentitis and necrosis of placental tissues [
79,
80]. The amount of
C. burnetii DNA detected increases until parturition and decreases drastically after parturition, probably by the disappearing of trophoblasts, the replication niche of
C. burnetii during pregnancy [
79,
81]. This strong tropism of
C. burnetii towards placenta does not seem to occur for other tissues of nonpregnant goats and kids, suggesting that pregnant females are more susceptible to
C. burnetii infection [
79,
82].
Cell-mediated immunity probably plays a critical role in controlling
C. burnetii infection [
49,
55]. Cells belonging to monocyte-macrophage lineage express polarized functional properties. This polarization seems to be closely related to the ability to control
C. burnetii infection, explaining the bacterial persistence in chronic infections [
83]. Classically, M1 polarized macrophages are induced by LPS, IFN-γ, and TNF-α, and participate in the resistance against intracellular pathogens involved in Th1 responses. In contrast, M2-polarized macrophages are induced by IL-4, IL-13, or IL-10 and promote Th2 responses. So, it is thought that the course of infection differs according to the macrophage polarization in response to
C. burnetii infection [
83]. If M1-associated molecules are expressed by macrophages, the bacterial replication will be controlled [
62,
83], while the stimulation of an M2 response will account for the persistence of
C. burnetii in macrophages, which become highly permissive to
C. burnetii replication [
83,
84,
85].
Beyond cell-mediated response, an antibody-mediated immunity also seems to be important in
C. burnetii infection [
49]. Treatment of
C. burnetii infection with immune sera makes the bacterium more susceptible to phagocytosis and destruction by macrophages [
86]. Specific immunoglobulins are secreted following infection [
38] and the infection of dendritic cells with antibody-opsonized bacteria results in increased expression of maturation markers and inflammatory cytokines in mice [
49]. It can be concluded from field studies that
C. burnetii antibodies are highly persistent, lasting for several months up to years [
87,
88]. Thus, both humoral and cellular immunity play a role in
C. burnetii infection.
However, the immune control of
C. burnetii might not lead to its eradication from the infected host [
55]. It is also hypothesized that the uterus could be a site of latent infection, hence reactivation during pregnancy can occur [
89,
90].
In goats, as well as in other domestic ruminants,
C. burnetii infection often goes unnoticed owing to the absence of symptoms, and the term Coxiellosis is usually used to refer this condition [
8]. In the early stages after infection,
C. burnetii can be detected in the blood, lungs, spleen, and liver. However, it is not clear if its presence in organs other than placenta affects the functions of these organs, as only mild lesions have been described [
79,
81,
91,
92]. Experimental infection of non-pregnant goats showed that, at late stages of infection,
C. burnetii was present in mammary glands, emphasizing the milk as an important shedding route [
82]. Infection of pregnant goats may cause a wide range of conditions including abortion, delivery of premature offspring, stillbirth, and weak offspring. Of these, one of the most important outcomes of the
C. burnetii infection is the abortion, which occur at the end of pregnancy without premonitory signs. In dairy goat herds that experience abortions caused by
C. burnetii, an increased incidence of metritis can be noticed. Notwithstanding, a clinically normal progeny, which may or may not be congenitally infected, may occur, as described in infection of non-pregnant goats [
7,
79,
82]. However, it seems that apparently healthy kids born from infected mothers may develop respiratory and digestive tract disorders [
7].
In the season that follows an abortion storm, the multiplication of the organism may be reactivated during pregnancy, leading to reproductive failures [
93,
94,
95]. Even in asymptomatic infections, a latent infection may develop and a reactivation late in pregnancy can occur several days before parturition. Generally, when late-term abortions, stillbirths, or birth of stunted animals are observed in goat flocks, Q fever should be suspected. Usually, up to 90% of the reproductive females within the flock are infected. This is why it is mentioned that
C. burnetii may cause epidemic herd outbreaks with significant animal losses owing to abortion waves and weak offspring during the parturition period [
96,
97].
This entry is adapted from the peer-reviewed paper 10.3390/biology11121703