Cranial and peripheral nerve sheath tumors (PNST) comprise a heterogeneous group of soft tissue tumors. Most arise from classic peripheral nervous system elements (Schwann cells and perineurial cells), while others involve specialized neuroendocrine cells of the sympathetic and parasympathetic nervous system (e.g., cauda equina neuroendocrine tumors, previously known as “CNS paragangliomas”). The World Health Organization (WHO) classification of the Central Nervous System (CNS) Tumors of 2021 and the 2020 WHO Classification of Soft Tissue and Bone Tumors include benign and malignant tumors, such as schwannoma, neurofibroma, plexiform neurofibroma (PN), perineurioma, hybrid nerve sheath tumor (HNST), malignant peripheral nerve sheath tumor (MPNST), epithelioid MPNST or malignant melanotic nerve sheath tumor (MMNST), and cauda equine neuroendocrine tumor. All these entities may arise along the craniospinal axis and be encountered either sporadically or as part of neurocutaneous syndromes, including neurofibromatosis type 1 (NF1), schwannoma predisposition syndromes (SPS), and Carney complex.
1. Epidemiology and Clinical Features
Schwannomas are benign nerve sheath tumors composed of differentiated neoplastic Schwann cells. Up to 90% of schwannomas are solitary and sporadic, and affect people of all ages, with a peak incidence in the fourth to sixth decades of life. An increased incidence of schwannomas after prior irradiation has been reported [
4]. A common presentation consists in an asymptomatic lesion of the skin or subcutaneous tissue of the head and neck, or along the flexor surfaces of the extremities. Spinal intradural extramedullary location is also frequent in schwannomas, that grow through neural foramina, causing radicular pain or sensory or, less frequently, motor symptoms. Spinal intramedullary and intracranial parenchymal schwannomas are rare [
5], as well as the involvement of abdominal viscera (e.g., gastrointestinal tract) or bone [
6]. Multiple paraspinal schwannomas are typical of neurofibromatosis type 2 (NF2). A frequent cranial location is the vestibular compartment of the eight cranial nerves. Vestibular schwannomas represent the third most common intracranial non-malignant tumor entity and comprise over 80% of tumors in the cerebellopontine angle [
7]. Bilateral involvement is a criterion for NF2 [
8]. Most of patients report unilateral hearing loss (94%) and tinnitus (83%), while vestibular symptoms, such as vertigo and unsteadiness, range from 17% to 75% of patients [
9]. Trigeminal and facial neuropathies, brainstem compression, and hydrocephalus may occur in case of large cerebellopontine angle tumors. Schwannomas do not usually recur if treated by gross total resection. Malignant transformation is exceptionally rare, and few case reports have reported a transformation in epithelioid MPNST [
10,
11,
12].
Neurofibroma is a frequent benign peripheral nerve sheath tumor (5.3% of all benign soft tissue tumors), and appears as a soft, skin-colored papule or small subcutaneous nodule. The most common site is the skin, with predominant dermal involvement; less frequently, the tumor involves medium-sized nerves, a nerve plexus, a major nerve trunk, or spinal nerve roots. Bilateral and/or multiple spinal root neurofibromas are typical of NF1, while cranial nerve neurofibromas are anectodical [
13]. Cutaneous neurofibromas are usually asymptomatic, rarely painful, and the most common chief complaint is the cosmetic appearance [
14]. Motor or sensory symptoms may occur when neurofibromas are in a deep location according with the distribution of the affected nerve. Neurofibroma can be classified into three types, such as localized or solitary, diffuse, and PN. Localized neurofibromas occur as polypoid lesions without any anatomical preference, while diffuse neurofibromas appear as plaque-like lesions mainly in the head and neck region. PN are large, massive lesions with “bag of worms” aspects, close to large spinal roots of the shoulder or pelvic girdle in adult population [
15], while craniofacial site with disfigurement (35%), or location along limbs (19%), in association with pain and impairment of function, are typical of childhood [
16]. The presence of multiple neurofibromas or PN is strongly suggestive of NF1, especially when associated with typical findings. PN with progressive growth in adolescents and young adults may be considered as premalignant lesions and called atypical neurofibromatous neoplasms of uncertain biological potential (ANNUBP), which need careful surveillance to prevent malignant degeneration [
17].
MPNST are rare aggressive tumors with an estimated incidence of 1.46 per 1,000,000 individuals [
19] accounting for 2–10% of all soft tissue sarcomas [
20]. Perineural MPNST are the most frequent form (93–95%), while epithelioid MPNST are particularly rare (~5% of all cases). Typically, MPNST arise from a pre-existing benign nerve sheath tumor, or deep-seated PN, or large intraneural neurofibroma, or ANNUBP in 8–13% of patients with NF1 [
21], representing almost 50% of all MPNST cases. Another 40% of MPNST occur sporadically, and 5% following radiotherapy [
22]. Patients with MPNST commonly are 20- to 50-year-olds and display large masses, primarily located along extremities, trunk, head, and neck area [
23], that may cause pain or other neuropathic symptoms [
18]. MPNST have a significant risk to recur (40–65%) and metastasize (40–80%) [
24,
25], resulting in a poor prognosis, with 5-year overall survival (OS) following treatments of 30–60% [
24,
26,
27].
Malignant melanotic nerve sheath tumors (MMNST) are rare aggressive neoplastic lesions with fewer than 200 cases reported thus far [
28]. MMNST occur mainly in young adults with a median age of 22 when associated with Carney complex (hereditary condition associated with spotty skin pigmentation, myxomas, and hormone-producing glands tumors), while in patients with sporadic presentation MMNST tend to occur later (median age of 33 years) [
28,
29]. Of note, the association between MMNST and Carney complex is not clearly established, with some series reporting >50% of patients with MMNST and Carney complex, and other showing an association in ≤5% only [
28,
30,
31].
Perineuriomas are slowly progressive peripheral nerve tumors arising from perineural cells, representing approximately 1% of peripheral nerve tumors [
36]. Most of them occur in major peripheral nerves and their branches, such as the sciatic nerve (42%), median nerve (13.5%), radial nerve (15.6%), and brachial plexus (12.2%). Uncommon locations are cranial nerves [
37,
38], lateral ventricle [
39], oral cavity, skin, and mandible [
40]. Most of patients present a focal, unilateral lesion, while only a few have been reported to present with bilateral (2.7%) or unilateral multifocal lesions (1.3%) [
40].
Hybrid nerve sheath tumors (HNST) are an extremely rare entity, characterized by hybrid lesions with mixed elements of neurofibroma, schwannoma, and/or perineurioma. Typically, HNST consists of a painless mass in subcutaneous tissue or dermis [
48]; when large peripheral nerves or spinal nerves are involved, the tumor may be associated with pain or neurological impairment. The most common site of HNST are the fingers [
49], but some rare cases arise from cranial nerves [
50,
51,
52]. Hybrid schwannomas/perineuriomas are sporadic [
52], while hybrid neurofibroma/schwannomas are strongly associated with NF1 and 2, and schwannomatosis [
53]. Hybrid neurofibroma/schwannoma is the most represented morphology (71%) in schwannomatosis [
53,
54], while hybrid neurofibroma/perineurioma has been reported in association with NF1 [
55,
56]. Overall, HNST are benign tumors with a rare propensity to recur locally.
2. Genetic Tumor Syndromes Correlated with Cranial and Peripheral Nerve Sheath Tumors
Some genetic tumour syndromes predispose to the development of central and PNST, such as NF1 and SPS. NF1 is an autosomal dominant disorder with a complete penetrance due to a germline pathogenic variant in the NF1 gene on 17q11.2, which is a down-regulator of the oncogene Ras. The prevalence of the disease is of 1/2000 to 1/4000 persons without any difference in incidence by sex or race. Phenotype may be heterogenous among members of the same family [
69]. Genetic mosaicism, resulting from somatic pathogenic variants in the NF1 gene after fertilization, represents 30–50% of “de novo” cases of NF1 and may cause anatomically limited or clinical attenuated phenotype. Several non-neoplastic manifestations may be present, including café au lait macules, skeletal abnormalities (scoliosis—often associated with paraspinal tumors, pseudarthrosis, early osteoporosis, long-bone dysplasia), short stature, vascular and congenital cardiac anomalies (renal artery stenosis, moyamoya syndrome, aneurysms), and macrocephaly. Learning disability and attention deficit/hyperactivity disorders occur in 50% or more of patients with NF1. PNST associated with NF1 are cutaneous neurofibromas (99%), PN (40–50%), MPNST (8–13%) in adult patients with NF1. Other tumors associated with NF1 are optic pathway gliomas, with the greatest risk prior to age of 6 years (15–20%), breast cancers, with greatest increase in risk between 30 and 40 years of age, pheochromocytomas (1–5%), GIST, and glomus tumors [
70]. Legius et al. [
71] have revised the major developments in genetics, ophthalmology, dermatology, neuroimaging, and provided an update on diagnostic criteria for NF1. Molecular diagnosis of NF1 is confirmed when an NF1 pathogenic variant is identified in an individual/fetus having either one or more of the other clinical diagnostic criteria. In fact, the identification of an NF1 variant alone does not allow a diagnosis of NF1, and requires further clinical and genetic evaluation [
71]. Dosage analysis for the identification of copy-number variants, and DNA-based sequencing, can identify a pathogenic variant in approximately 90% of classic NF1 patients (e.g., with pigmentary features, as well as neurofibromas). Detection rate and specificity are increased to 95–97% when an RNA-based sequencing, in addition to dosage analysis, is applied [
72]. Mosaicism is confirmed when a patient with features of NF1 carries a heterozygous NF1 pathogenic variant with a variant allele fraction (VAF) < 50% in an unaffected tissue (e.g., blood). Mosaicism is also confirmed if an identical pathogenic NF1 variant is identified in two or more anatomically unrelated lesions in the absence of this pathogenic variant in unaffected tissue such as blood [
71].
Neurofibromatosis type 2 (NF2) and schwannomatosis (SWN) are genetically distinct tumor predisposition syndromes with overlapping phenotypes, of whom diagnostic criteria have been recently updated. A diagnosis of NF2 can be made in case of bilateral vestibular schwannomas, presence of an identical NF2 pathogenic variant in at least two anatomically distinct NF-2-related tumors (e.g., schwannoma, ependymoma, meningioma) or when two major criteria or one major plus two minor criteria are met (major criteria: unilateral vestibular schwannoma, first-degree relative other than sibling with NF2, ≥2 meningiomas, NF2 pathogenic variant in unaffected tissue—if VAF < 50%, the diagnosis is mosaic NF2; minor criteria: more than one type of tumors, including ependymoma, meningioma, or schwannoma, juvenile subcapsula or cortical cataract, retinal hamartoma, epiretinal membrane in a person < 40 years) [
74].
SWN is a clinical distinct entity as a result of germline NF2 (22q-related schwannomatosis), or SMARCB1 (SMARCB1-related schwannomatosis), or LZTR1 variant (LZTR1-related schwannomatosis), which are located centromeric to NF2 on chromosome 22 [
74,
77]. Germline SMARCB1 or LZTR1 pathogenic variant account for 70–80% of familiar SWN, but 30% only of sporadic cases [
77]. Major criteria for the diagnosis of SWN are at least one pathologically confirmed schwannoma or HNST and an SMARCB1 or LZTRK1 pathogenic variant in unaffected tissue (e.g., blood) or a shared SMARCB1 or LTRK1 pathogenic variant in two schwannomas or HNST [
74]. As several tumor types and clinical features are shared by NF2 and SWN, a patient suspected for one of these SPS should undergo comprehensive genetic testing to achieve the correct diagnosis.
3. Pathology and Molecular Markers
Tumor diagnosis for most of cranial and paraspinal nerve sheath tumors is still primarily based on hematoxylin and eosin (H&E)-stained sections and some additional techniques, including immunohistochemistry. Molecular testing generally is not required for this type of tumor but may be of help in the distinction of low-grade MPNST from cellular or atypical neurofibroma. However, mutation analysis of PNST may be required to diagnose mosaic forms of NF1, NF2, and SPS through the identification of the same mutation in at least two independent tumors as this is often the only way to prove a mosaicism.
Schwannoma. Conventional schwannoma is an encapsulated tumor comprised almost exclusively of neoplastic Schwann cells arranged in an alternating pattern of hypercellular Antoni A areas and hypocellular Antoni B areas. Nuclear palisading may be present, and Verocay bodies may be seen. The stroma can show hyalinized blood vessel walls and foamy macrophages. Rare histological variants include cellular, plexiform, microcystic, reticular, and epithelioid schwannomas. Tumors of the eighth cranial nerve are unencapsulated and predominantly show Antoni A tissue. Ancient schwannoma differs from conventional schwannoma only by its presence of scattered atypical to bizarre-appearing nuclei. A malignant change of schwannoma is exceptionally rare [
1,
2]. The tumour cells are diffusely and strongly positive for S100 protein, with both nuclear and cytoplasmic staining and show uniform nuclear positivity for SOX10 [
78,
79]. Loss of SMARCB1 (INI1) expression is found in epithelioid schwannoma, or a mosaic pattern of SMARCB1 (INI1) expression indicates syndrome-associated schwannoma [
80,
81]. Schwannomas may exhibit complete or partial loss of chromosome 22. Despite frequent NF2 alterations in schwannomas, this is not specific, and a pathognomonic molecular signature has not been found. However, schwannomas exhibit a distinct DNA methylation pattern [
1,
2].
Neurofibroma and plexiform neurofibroma. Neurofibromas show a diffuse proliferation of Schwann cells, nerve sheath fibroblasts, and less markedly axons permeating the lesion in a haphazard fashion in a myxoid and collagenous stroma. Furthermore, neurofibromas show a disperse positive staining for S-100 of only a portion of the tumor cells. An increased cellularity in the absence of other features may be seen in cellular neurofibromas. Mitotic figures are not usually present and may denote an atypical neurofibroma (AN) or atypical neurofibromatous neoplasm of uncertain biological potential (ANNUBP) in the setting of NF1. Malignant transformation requires a triad of increased cellularity, nuclear pleomorphism, and increased mitotic figures. The current WHO classification provides exact criteria for NF vs. ANNUBP and MPNST [
1,
2]. Localized cutaneous neurofibromas are consistently benign, while PN, ANNUBP, and solitary intraneural neurofibromas arising in sizeable nerves can be precursor lesions of MPNST. A biallelic genetic inactivation of the tumor suppressor gene NF1 is almost invariably present in NF [
82]. Histological features of AN/ANNUBP are strongly associated with deletions of the CDKN2A/CDKN2B locus, which may be demonstrated immunohistochemically by p16 loss [
2]. Inactivation of SUZ12 or EED, leading to H3K27 trimethylation loss, denotes progression to MPNST [
2].
Perineurioma. Perineurioma is composed of spindle cells with wavy or tapering nuclei, indistinct nucleoli, and bipolar cytoplasmic processes arranged in a storiform or whorled growth pattern. The intraneural lesions form typical pseudo-onion bulbs. Tumor cell stain is variably positive for EMA, claudin-1, and GLUT1. CD34 is expressed in 60% of cases. The cells are negative for S100 and SOX10 [
1,
2]. Intraneural perineuriomas harbor missense mutations in TRAF7 [
83], whereas soft tissue perineuriomas commonly show deletions of chromosome 22q (NF2) and deletions of chromosome 17q11 (NF1) [
84], as well as chromosome 2p deletions or rearrangements or deletions of chromosome 10q (sclerosing variant) [
85].
Hybrid nerve sheath tumors. HNST are benign PNST with combined features of more than one conventional type (neurofibroma, schwannoma, perineurioma) [
1,
2]. The molecular features of the dual differentiation, which is typical of hybrid tumors, are largely unknown. Activating ERBB2 mutations have been identified in a subset of neurofibroma/schwannoma hybrid tumors [
86].
Malignant peripheral nerve sheath tumor (MPNST). Morphologic criterion for the diagnosis of MPNST is the presence of spindle cells with indistinct cytoplasmic margins and wavy or S-shaped nuclei, that are arranged in fascicles with alternating cellular and myxoid areas. Brisk mitotic activity is usually present. Rare dispersed, single-tumor cells stain positive for S100 in about 65% of tumors [
1,
2]. Most of MPNST show homozygous inactivation of NF1 and CDKN2A and/or CDKN2B. Inactivation of the core components of PRC2, SUZ12, or EED is the most important molecular marker for MPNST [
87,
88,
89,
90]. This leads to loss of H3K27 trimethylation, which may be demonstrated by immunohistochemistry [
91,
92].
4. Imaging
Imaging is used to orient the diagnosis, delineate lesion margins and involved structures, and monitor central and peripheral nerve sheath tumors. Ultrasounds should be the first-line imaging procedure in the presence of a superficial musculoskeletal soft-tissue lesion. This affordable, radiation-free technique allows an initial exclusion of mimicking pathologies with high diagnostic accuracy [
96,
97] and can guide percutaneous biopsy with a reasonably low complication rate [
98,
99]. Computerized tomography (CT) should not be used to characterize neurogenic lesions, as it displays low contrast resolution in soft tissue structures and exposes patients to ionizing radiation. However, CT can detect bone remodeling and/or erosion [
100], hemorrhagic transformation in emergency settings [
101], and calcifications [
102].
Magnetic resonance imaging (MRI) is the gold standard imaging modality to characterize soft tissue lesions [
103,
104] and delineate their extension to adjacent structures (especially neurovascular structures and muscular fascia). MRI can non-invasively suggest the neurogenic origin of a soft tissue mass and may help to distinguish between benign and malignant variants. However, MRI is limited in the distinction between schwannoma and neurofibroma [
105], as well as between schwannoma and MPNST [
106,
107]. The MR-imaging-based Neuropathy Score Reporting and Data System (NS-RADS) is a recently developed framework to standardize MRI reporting in peripheral neuropathy [
108,
109].
Positron emission tomography (PET) combined with CT is useful in detecting malignant transformation of a PNST, and identifying systemic metastases, and thus guiding biopsy [
110]. F18 fluorodeoxyglucose (FDG) is the most used PET tracer. Early and delayed (60–90 min and 240 min, respectively) FDG-PET/CT scans diagnose NF1-associated MPNST with high (~90%) sensitivity and specificity values [
106]. Benign lesions display low (<2–3) maximum standardized uptake value (SUVmax) on early and delayed FDG-PET scans, while MPNST show increased (>3–4) values. PET/MRI has been suggested as a feasible alternative to PET/CT in patients with NF1 when screening for presence of MPNST. The main benefit is to avoid the exposure to ionizing radiations from CT with similar accuracy (100%) of PET/MRI to detect MPNST as compared with PET/CT [
111,
112]. 11C-methionine tracer increases PET specificity in ambiguous lesions [
113].
MRI features of a neurogenic tumor. The direct continuity between a nerve and a lesion, resembling a tail coming off the lesion, is called a tail sign [
106,
107]. This feature is almost pathognomonic of a PNST, particularly when seen along the long axis of a lesion and when a large nerve is involved. However, MRI does not discriminate between benign and malignant conditions. The tail sign is more commonly located in the central part of the lesion in neurofibromas, while it is more eccentrically located in schwannomas.
MRI features of benign PNST. Several findings have been described in benign PNST, including a tail sign, fusiform shape, well-defined margins, a target sign (a low or intermediate signal intensity in the center of the lesion, surrounded by a peripheral hyperintense ring), a fascicular sign (various thin ring-like structures), and a split fat sign (a fat rim that separates the tumor from the surrounding tissue) [
107,
114,
115]. Wide window settings allow a better detection of most of these imaging characteristics. Lesions are usually T1 hypointense, T2 hyperintense, and display a significant Gadolinium enhancement, which is delayed on dynamic contrast-enhanced imaging. Fat suppressed T2 and T1 sequences after contrast injection are often the most helpful to show the lesion.
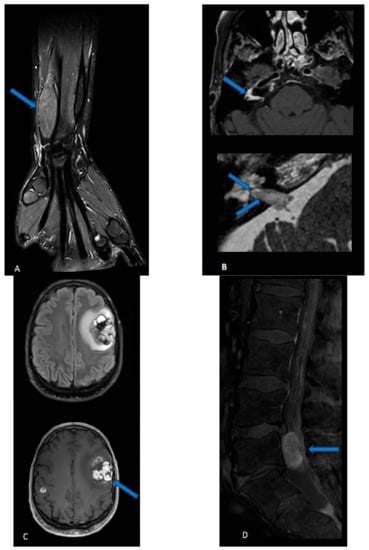
Perineuriomas are seen as a gradual, uniform nerve enlargement, followed by a gradual narrowing (
Figure 1A). Individual fascicles are uniformly enlarged, displaying a characteristic “honeycombing pattern” [
106]. Increased apparent diffusion coefficient (ADC) values (>1.1 × 10
−3 mm
2/s) and functional anisotropy (FA) values are also seen [
116]. On MRI spectroscopy, the trimethylamine fraction is usually low (<50%) [
117]. Bone destruction is not a specific feature in the distinction between benign and MPNST [
118]. The radiological features of HNST are lacking: a single case report described multiple nerve sheath lesions with a bright signal on STIR sequence, peripheral enhancement, and large avascular regions [
119].
Figure 1. (A) Perineurioma: coronal three-dimensional (3D) proton density sequence of the wrist after fat suppression displays a fusiform enlargement of the radial nerve. (B) Malignant peripheral nerve sheath tumor: Three-dimensional T1-weighted spin echo sequence after contrast injection and fat suppression (upper image) shows a contrast-enhanced lesion in the right internal auditory canal and geniculate ganglion. High-resolution 3D T2-weighted sequence (lower image) reveals intra-cochlear extension of the tissue mass. (C) Malignant intracerebral nerve sheath tumor: 3D FLAIR sequence in the axial plane (upper image) and 3D T1-weighted spin echo sequence after contrast injection shows two intra-axials, enhancing lesions. The biggest lesion, located in the left superior frontal region, is highly heterogeneous, with hemorrhagic and cystic components, and is surrounded by FLAIR hyperintensity. The second, smaller lesion of the right hemisphere is hardly visible on the FLAIR image while it strongly enhances after contrast administration. (D) Cauda equine neuroendocrine tumor: sagittal T1-weighted sequence after contrast injection and fat suppression of the lumbar spine displays a well-defined intradural extramedullary contrast, enhancing lesions.
MRI features of MPNST (
Figure 1B). If typical findings of a benign PNST are absent, an MPNST should be suspected. The presence of infiltrative margins, peritumoral edema and/or necrosis, intra- or peritumoral hemorrhage, an irregular or round shape, a size greater than 5 cm, and a heterogeneous enhancement all suggest a malignant tumor [
120]. ADC values are low (<1.1
× 10
−3 mm
2/s), nerve tracts are partially or completely disrupted in diffusion tensor studies, and trimethylamine fraction is high (greater than 50%) in MPNST. In the case of neurofibromatosis or in the follow-up of a known benign PNST, a rapid growth is also evocative of malignant degeneration [
121]. Of note, it can be difficult to establish malignancy on imaging, and some radiological findings (heterogeneity, diffusion restriction) are less concerning in schwannoma in comparison to neurofibroma and plexiform neurofibroma. MPNSTs have a strong tendency to metastasize, especially in the spinal canal, and therefore spine imaging should be performed. In the neuroaxis, extra-axial MPNSTs are commonly separated from their intraparenchymal counterparts, which are termed malignant intracerebral nerve sheath tumors (MINST) (
Figure 1C) [
122,
123]. MINST display nonspecific high-grade tumor features (high heterogeneity, variable necrotic, hemorrhagic, cystic and calcific components, irregular enhancement) and therefore are indistinguishable from a high-grade glial neoplasm or a solitary metastasis.
5. Surgery
The vast majority of PNST is benign and well-circumscribed; therefore, neurosurgical resection is the therapy of choice not only to gain tissue for the diagnosis but also to cure the tumor [
131,
132]. Principles of surgery in the different entities of tumors involving peripheral nerves, such as schwannoma, neurofibroma, perineurinoma, HNST, MPNST, and cauda equina neuroendocrine tumors, are similar, but might differ depending on tumor size, location, attachment to neighboring structures, and malignancy. Basically, total removal of the tumor is the goal of surgery. However, the extent of surgery must be balanced to preservation of nerve function. Complete resection can be achieved by intracapsular dissection of the tumor mass, preserving the attached functional nerve fibers [
131,
132,
133]. Intraoperative electrophysiology, especially direct motor nerve stimulation, enables the neurosurgeon to detect functional nerve fibers, even if they are displaced by the tumor, and to monitor nerve function during surgery. Therefore, the use of intraoperative electrophysiological monitoring is regarded as mandatory [
131,
132].
Cranial nerve tumors, such as vestibular schwannomas (VS), require a careful evaluation for surgery. Observing VS with serial MRI and audiological monitoring without any tumor-directed treatment is considered appropriate for incidental, asymptomatic VS [
137]. However, approximately 50% of VS may be expected to grow over a 5-year period [
138,
139] with a risk of 50% to lose functional hearing during a 3–4-year period [
140].
6. Radiotherapy
Radiotherapy is a central pillar in the multimodality treatment of soft tissue sarcomas and CNS tumors, also of mesenchymal origin like meningiomas and vestibular schwannomas. Depending on individual patient and tumor situation, it can be applied either as sole definitive treatment with curative intent or as pre- or post-operative adjunct to surgery to maximize local control in difficult-to-treat tumors or with palliative intent to alleviate symptomatic burden of the disease. The current standard of treatment planning and delivery is the use of multi-modality co-registered imaging for target volume definition, intensity modulated radiotherapy—either delivered as step-n-shoot or volumetric rotational radiotherapy—with integrated online image guidance. Currently, standard fractionation is mostly utilized with single fraction doses of 1.8–2.0 Gy up to total doses of 45–66 Gy, depending on the decision on pre- or post-operative radiotherapy. In case of definitive radiotherapy, stereotactic radiotherapy with single-fraction or multi-fraction radiosurgery has evolved as an alternative to conventional fractionated radiotherapy, especially for small volumes which are distant to critical organs where small safety margins can be applied. As there is no prospective (randomized) data available on the optimal treatment of CNS and peripheral nerve tumors, reliance on retrospective studies and case series, as well as on analogy assumptions from soft tissue sarcoma and CNS tumor management, is sensible.
Malignant nerve sheath tumors. Due to its aggressive nature and tendency for local relapse, radiotherapy has been considered for high-grade or large (>5 cm) tumors as well as after incomplete (R1 or R2) resection with a trend for improved local control [
145], but was not associated with improved overall survival as suggested by a recent nation-wide registry analysis [
146]. With the availability of the prospective study on non-rhabdomyosarcoma soft tissue sarcomas in patients under 30 years of age (COG study ARST0332; 11% of MPNST included), some guidance can be derived given the encouraging results, although generalization to older patients remains open [
147]; only in patients with low-grade tumors and gross total resection (R0 and R1) radiotherapy had been omitted.
Neurofibroma/Perineuroma/Hybrid nerve sheath tumors. Although these tumors comprise rare and different cohorts of benign tumors and prospective evidence is missing, some cautious conclusions can be drawn from the experience of treating vestibular schwannomas. Firstly, if tumors can be easily removed, patients should be advised to undergo surgery. If this is not possible and the patient is symptomatic or symptomatic progression with significant impact on quality of life is expected, radiotherapy should be explored. In small tumors less than 15 mm in size, single-fraction radiosurgery with 12–14 Gy is recommended, while conventionally fractionated stereotactic radiotherapy with 1.8 Gy to 50.4 Gy is applied in larger tumors, especially in areas near critical organs at risk.
7. Medical Treatments
7.1. Vestibular Schwannomas
Bevacizumab is the sole compound that has been successfully used for patients with progressive VS associated with NF2 with remarkable improvement of hearing and objective radiographic responses [
155]. Blakeley et al. have conducted a multi-institutional uncontrolled phase 2 study using 7.5 mg/kg bevacizumab administered every 3 weeks in NF2 patients with progressive VS, displaying a hearing improvement in 36% of patients, and a partial radiographic response with volume reduction of 20% or more in 43% (6/14 patients), making bevacizumab a potential treatment option for NF2 patients [
156].
7.2. Plexiform Neurofibromas
The discovery of the molecular pathogenesis of PN of neurofibromatosis has launched several clinical trials that have investigated targeted therapies. Some initial studies with imatinib (RTK inhibitor of the downstream pathways including MAPK, PI3K/AKT, and JAK/STAT) [
157], tipifarnib (RAS inhibitor) [
158], pirfenidone (TGF-β inhibitor) [
159], sirolimus (mTOR inhibitor) [
160,
161], interferon alfa-2b inhibitor [
162], and everolimus [
163] reported limited benefits. Conversely, the development of MEK inhibitors represents the first effective targeted therapy for PN [
164]. In this regard, selumetinib is an oral, highly potent, and selective MEK1 inhibitor, that has been investigated primarily in children with remarkable results in terms of disease control and quality of life. In a phase 1 trial on children and adolescents (3–18 years) with NF1 and inoperable PNs, the objective response rate (ORR) was of 71% (15/17 partial response -PR-, and 2 stable disease -SD-) with a median reduction in tumor volume of 31%.
Mirdametinib is an oral, highly selective small-molecule inhibitor of MEK1 and 2, that has been investigated in 19 patients aged ≥ 16 years with NF1 and progressive and/or symptomatic PNs, showing 2% of PR only, a median reduction in tumor volume of 17.1%, and an improvement of patient-reported outcomes over treatment. The most frequent AEs were acneiform rash, fatigue, and nausea [
171]. A further evolution of MEK1-2 inhibitors is represented by trametinib and binimetinib. The interim analysis of the phase 1–2 trial on trametinib in pediatric patients with NF-1–associated PN reported a PR in 12 out of 26 patients (46%) with long-lasting radiological response more than 12 months [
172] and manageable side effects, such as paronychia and rash.
7.3. Malignant Nerve Sheath Tumors
Given the high risk to progress locally and/or with systemic metastases, adjuvant treatments should be considered in patients who underwent surgery for MPNST regardless of the extent of resection. However, there is still controversy regarding which treatment modality (radiotherapy vs. systemic chemotherapy) and timing (neoadjuvant vs. adjuvant setting) is more effective, due to the lack of definitive data regarding either local disease control or decrease of risk of systemic progression [
26]. The key point for the treatment decision is the determination of recurrence risk and prognosis, which is mainly based on different factors, such as large size, high-grade histology, positive margins following resection, presence of necrosis, association with NF1 or previous radiation therapy [
178].
Traditional chemotherapy. In general, anthracycline-based treatment is the first-line therapy for unresectable, locally advanced, or metastatic MPNST. The choice of using single-agent anthracycline (doxorubicin) or combined treatment (doxorubicin/ifosfamide) depends on patient-specific factors (i.e., performance status, comorbidities) since the combination of doxorubicin/ifosfamide increases both radiological responses and toxicity, but not OS as compared with single agent doxorubicin [
179]. A pooled analysis of 12 EORTC- Soft Tissue and Bone Sarcoma Group (STBSG) trials suggested a minimal superiority of the doxorubicin/ifosfamide regimen over doxorubicin monotherapy in MPNST with median PFS (17 vs. 16 weeks), but not for median OS (48 vs. 51 weeks) [
180]. In case of progression following anthracycline-based therapy, other regimens include the topoisomerase II inhibitor etoposide based on the rationale that the topoisomerase-IIα is overexpressed in MPNST [
181]. In fact, the SARC006 trial reported that 5/48 patients with MPNST had tumor shrinkage after two cycles of etoposide/ifosfamide after progression following doxorubicin/ifosfamide [
182]. Moreover, a case series showed two patients with MPNST with significant responses to etoposide/carboplatin after refractory disease to doxorubicin/ifosfamide [
183]. Further cytotoxic chemotherapy regimens can be considered when anthracycline and etoposide-based therapy fails to control the disease; however, limited data on their efficacy have been reported thus far. For instance, gemcitabine in combination with docetaxel was delivered in two patients with MPNST reporting a PR [
184], as well as in Japanese retrospective series showed a stabilization of disease in four out of five patients [
185]. Last, other compounds, such as carboplatin, dactinomycin, cisplatin, vincristine, cyclophosphamide, imidazole, and carboxamide, have been employed, alone or in combination, for the treatment of MPNST, with disappointing results [
186].
8. Conclusions
Surgery is the therapy of first choice in central and peripheral nerve sheath tumors to obtain a histological diagnosis and reduce tumor burden with the primary aim to preserve surrounding soft tissues and nerve functioning. In this regard, gross total resection may be curative for benign tumors. Additionally, the extent of resection is crucial for reducing residual tumors and is correlated with OS in MPNST. Given the heterogeneity and rarity of these tumors, there is a paucity of well-powered clinical trials, thus it is not possible to generate evidence-based treatment recommendations for non-surgical modalities. However, some clinical trials have been reported on targeted therapies in plexiform neurofibromas of NF1 patients or in heterogenous cohorts of soft-tissue tumors, including MPNSTs, with initial data of efficacy that need to be further investigated.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15071930