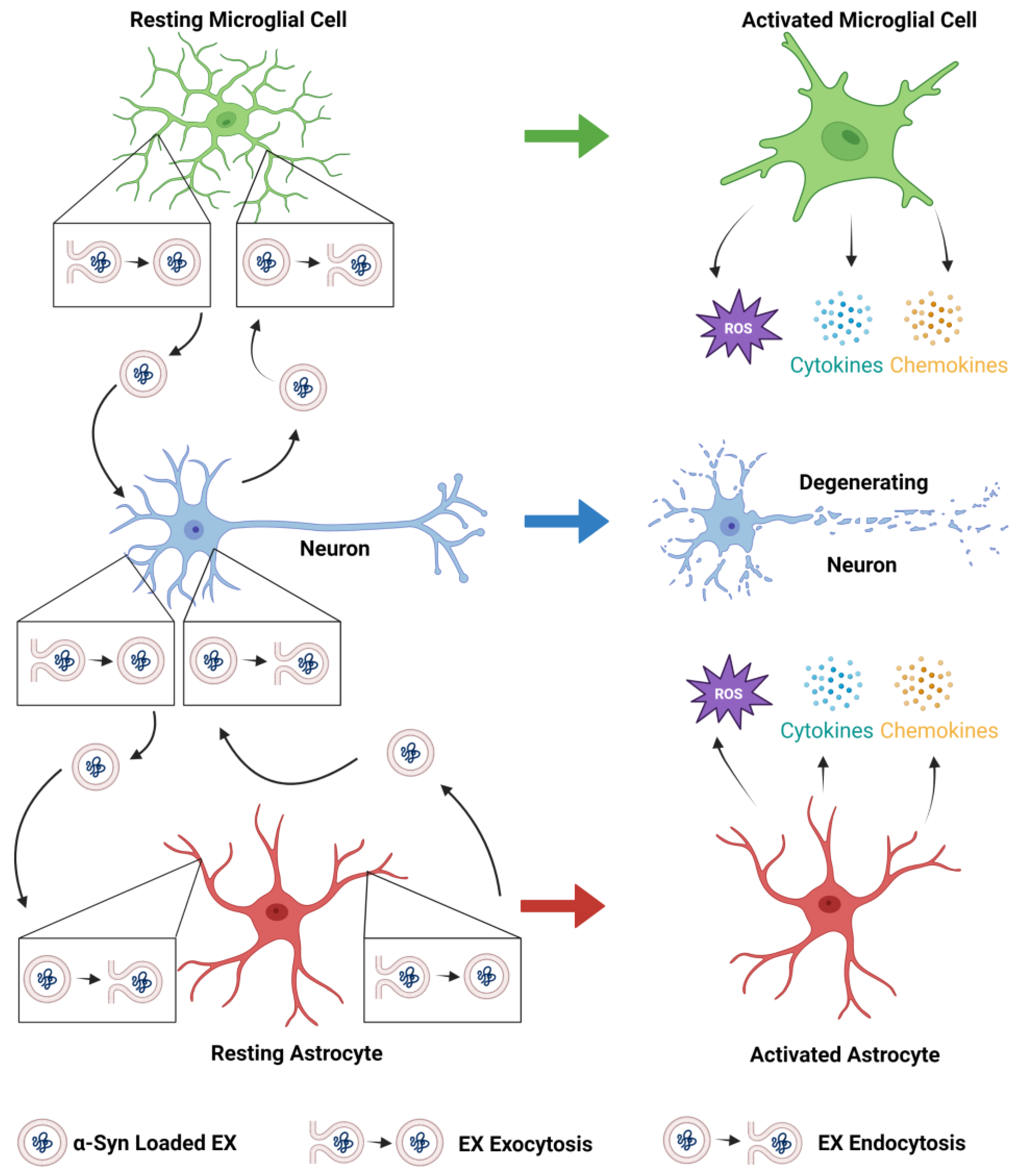
Extracellular vesicles (EVs) are membrane-enclosed particles released by cells into the extracellular space. EVs can be classified as exosomes (EXs), microvesicles, or apoptotic bodies based on their origin and size. EXs are enclosed within a single phospholipid bilayer, secreted by all cell types, formed by the inward invagination of the endosomal membrane and fusion of the multivesicular body (MVB), and are typically 30–150 nm in diameter. Microvesicles are EVs that form from direct outward budding from the cell’s plasma membrane and are typically 100 nm to 1 µm in diameter. Pathogenic forms of α-synuclein (α-syn) are transferred to and from neurons, astrocytes, and microglia, which spread α-syn pathology in the olfactory bulb and the gut and then throughout the Parkinson’s disease (PD) brain and exacerbate neurodegenerative processes.
- exosomes
- extracellular vesicles
- Parkinson’s disease
- pathogenesis
- therapeutics
- α-synuclein
1. Role of Exosomes in the Pathogenesis of Parkinson’s Disease
2. Pathogenic α-Syn-Containing Exosomes as Therapeutic Targets
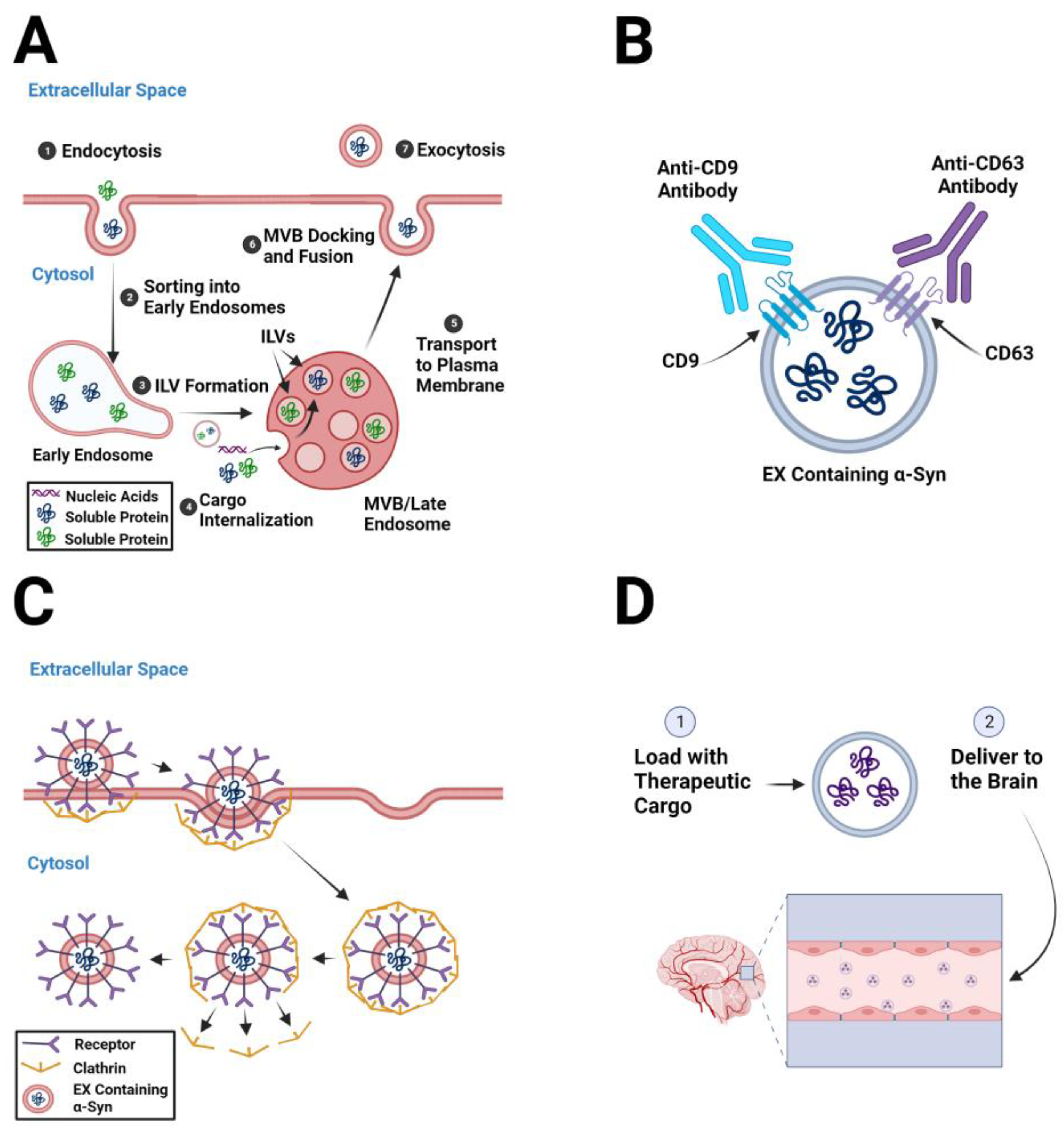
2.1. Decreasing Exosome Biogenesis
2.2. Depleting Circulating Pathogenic Exosomes
2.3. Inhibiting Exosome Uptake by Recipient Cells
3. Exosomesas Therapeutic Delivery Systems in Parkinson’s Disease
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11041187
References
- Lebouvier, T.; Chaumette, T.; Paillusson, S.; Duyckaerts, C.; Bruley des Varannes, S.; Neunlist, M.; Derkinderen, P. The second brain and Parkinson’s disease. Eur. J. Neurosci. 2009, 30, 735–741.
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Park. Dis. 2018, 4, 21.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.; National Institute for Clinical, E. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245.
- Liu, W.; Zhang, Q.; Xing, H.; Gao, G.; Liu, J.; Huang, Y.; Yang, H. Characterization of a Novel Monoclonal Antibody for Serine-129 Phosphorylated alpha-Synuclein: A Potential Application for Clinical and Basic Research. Front. Neurol. 2022, 13, 821792.
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841.
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169.
- Ibanez, P.; Bonnet, A.M.; Debarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Durr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576.
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004, 576, 363–368.
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966.
- Chu, Y.; Kordower, J.H. The prion hypothesis of Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2015, 15, 28.
- Kordower, J.H.; Brundin, P. Propagation of host disease to grafted neurons: Accumulating evidence. Exp. Neurol. 2009, 220, 224–225.
- Olanow, C.W.; Brundin, P. Parkinson’s disease and alpha synuclein: Is Parkinson’s disease a prion-like disorder? Mov. Disord. 2013, 28, 31–40.
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024.
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367.
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell Responses to Extracellular alpha-Synuclein. Molecules 2019, 24, 305.
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67.
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947.
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of alpha-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982.
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015.
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725.
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851.
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 46.
- Burbidge, K.; Rademacher, D.J.; Mattick, J.; Zack, S.; Grillini, A.; Bousset, L.; Kwon, O.; Kubicki, K.; Simon, A.; Melki, R.; et al. LGALS3 (galectin 3) mediates an unconventional secretion of SNCA/alpha-synuclein in response to lysosomal membrane damage by the autophagic-lysosomal pathway in human midbrain dopamine neurons. Autophagy 2022, 18, 1020–1048.
- Han, C.; Xiong, N.; Guo, X.; Huang, J.; Ma, K.; Liu, L.; Xia, Y.; Shen, Y.; Li, J.; Jiang, H.; et al. Exosomes from patients with Parkinson’s disease are pathological in mice. J. Mol. Med. 2019, 97, 1329–1344.
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229.
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428.
- Liu, J.; Zhou, Y.; Wang, Y.; Fong, H.; Murray, T.M.; Zhang, J. Identification of proteins involved in microglial endocytosis of alpha-synuclein. J. Proteome Res. 2007, 6, 3614–3627.
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 2020, 143, 1476–1497.
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195.
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641.
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208.
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47.
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685.
- Song, L.; Tang, S.; Han, X.; Jiang, Z.; Dong, L.; Liu, C.; Liang, X.; Dong, J.; Qiu, C.; Wang, Y.; et al. KIBRA controls exosome secretion via inhibiting the proteasomal degradation of Rab27a. Nat. Commun. 2019, 10, 1639.
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232.
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12 (Suppl. S11–S13), 19–30.
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Thery, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930.
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244.
- Jiang, T.; Xu, C.; Gao, S.; Zhang, J.; Zheng, J.; Wu, X.; Lu, Q.; Cao, L.; Yang, D.; Xu, J.; et al. Cathepsin L-containing exosomes from alpha-synuclein-activated microglia induce neurotoxicity through the P2X7 receptor. NPJ Park. Dis. 2022, 8, 127.
- Tsutsumi, R.; Hori, Y.; Seki, T.; Kurauchi, Y.; Sato, M.; Oshima, M.; Hisatsune, A.; Katsuki, H. Involvement of exosomes in dopaminergic neurodegeneration by microglial activation in midbrain slice cultures. Biochem. Biophys. Res. Commun. 2019, 511, 427–433.
- Zhu, C.; Bilousova, T.; Focht, S.; Jun, M.; Elias, C.J.; Melnik, M.; Chandra, S.; Campagna, J.; Cohn, W.; Hatami, A.; et al. Pharmacological inhibition of nSMase2 reduces brain exosome release and alpha-synuclein pathology in a Parkinson’s disease model. Mol. Brain 2021, 14, 70.
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191.
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250.
- Kawamoto, T.; Ohga, N.; Akiyama, K.; Hirata, N.; Kitahara, S.; Maishi, N.; Osawa, T.; Yamamoto, K.; Kondoh, M.; Shindoh, M.; et al. Tumor-derived microvesicles induce proangiogenic phenotype in endothelial cells via endocytosis. PLoS ONE 2012, 7, e34045.
- Lima, L.G.; Chammas, R.; Monteiro, R.Q.; Moreira, M.E.; Barcinski, M.A. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009, 283, 168–175.
- Plebanek, M.P.; Mutharasan, R.K.; Volpert, O.; Matov, A.; Gatlin, J.C.; Thaxton, C.S. Nanoparticle Targeting and Cholesterol Flux Through Scavenger Receptor Type B-1 Inhibits Cellular Exosome Uptake. Sci. Rep. 2015, 5, 15724.
- Weiss, J.L.; Ng, L.K.; Chase, T.N. Long-lasting dyskinesia induced by levodopa. Lancet 1971, 1, 1016–1017.
- Kang, U.J.; Fahn, S. Management of tardive dyskinesia. Ration. Drug Ther. 1988, 22, 1–7.
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Fox, S.H.; Brotchie, J.M. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 2013, 65, 171–222.
- Kuwabara, H.; Cumming, P.; Yasuhara, Y.; Leger, G.C.; Guttman, M.; Diksic, M.; Evans, A.C.; Gjedde, A. Regional striatal DOPA transport and decarboxylase activity in Parkinson’s disease. J. Nucl. Med. 1995, 36, 1226–1231.
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513.
- Gomez-Molina, C.; Sandoval, M.; Henzi, R.; Ramirez, J.P.; Varas-Godoy, M.; Luarte, A.; Lafourcade, C.A.; Lopez-Verrilli, A.; Smalla, K.H.; Kaehne, T.; et al. Small Extracellular Vesicles in Rat Serum Contain Astrocyte-Derived Protein Biomarkers of Repetitive Stress. Int. J. Neuropsychopharmacol. 2019, 22, 232–246.
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnol. 2018, 16, 81.
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166.
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30.
- Vilaca-Faria, H.; Salgado, A.J.; Teixeira, F.G. Mesenchymal Stem Cells-derived Exosomes: A New Possible Therapeutic Strategy for Parkinson’s Disease? Cells 2019, 8, 118.
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564.
- Lee, E.J.; Choi, Y.; Lee, H.J.; Hwang, D.W.; Lee, D.S. Human neural stem cell-derived extracellular vesicles protect against Parkinson’s disease pathologies. J. Nanobiotechnol. 2022, 20, 198.