1.1. Telomeres
Telomeres are nucleus-protein complexes located at the ends of the chromosomes of eukaryotic cells [
7]. This structure prevents the linear ends of chromosomes from being recognized as double-strand DNA breaks(DSBs) [
8]; their shortening constitutes a mechanism of tumor suppression by activating cellular senescence signals [
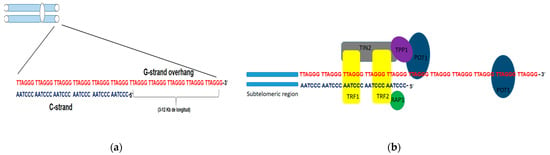
9]. In humans, telomeric DNA is composed of tandem repeats of the hexanucleotide 5′-TTAGGG-3′sequence. This sequence extends from 3 to 12 kb in length, giving rise to a single chain rich in guanine residues (G strand) (
Figure 1a). The protruding G chain can bend and invade the telomere double chain region and generate a loop structure, the T-loop, thathides the 3′end, as a primitive mechanism for telomere protection [
10].
Figure 1. Structure of telomeres in humans. (a) Schematic representation of telomeric DNA in humans, composed of tandem repeats of the hexanucleotide sequence TTAGGG (G chain) and its complementary sequence in chain C. The G chain can extend 3–12kb in length (G-strand overhang). (b) Adjacent to the telomeres are the sub telomeric regions, also rich in repetitive DNA. Schematic representation of the shelterin or telosome complex. The TRF1 and TRF2 proteins have a binding domain for double-stranded DNA, whereas POT1 can only bind to single-stranded DNA. RAP1 exerts its function on the telomere through a TRF2-binding domain. TIN2 has binding domains for both TRF1 and TRF2, and through another domain, it binds to the TPP1-POT1 complex.
Associated with these tandem repeats is a six-protein complex called shelterins, including TRF1, TRF2, TIN2, RAP1, TPP1, and POT1 (
Figure 1b). TRF1 and TRF2 contain a binding domain for dsDNA, while POT1 interacts with telomeric DNA. The other protein complex components do not have binding domains for telomeric DNA and exert their function through interaction with the TRF1 and TRF2 proteins. Shelterins prevent the DNA damage response mechanisms that DSBs activate from being triggered and in this way, prevent the occurrence of homology-directed repair (HDR) or binding processes non-homologous end joining (NHEJ). Likewise, TRF2 protein plays a key role in telomere protection since it makes the construction of the T-loop possible, giving telomeric DNA stability and inaccessibility at its ends. Therefore, telomeres devoid of shelterins lead to the fusion of the ends of chromosomes recognized as DSB that need to be repaired [
11,
12]. Even though telomeric shortening occurs after each cell cycle, other factors, such as oxidative stress, can influence the speed of telomeric shortening [
13,
14].
1.2. Mechanism Involved in Lengthening of Telomeres
Telomerase
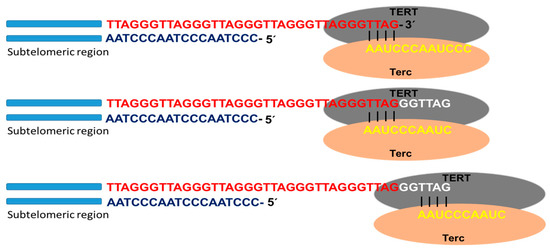
Telomerase is the specialized DNA polymerase responsible for the maintenance and elongation of telomeres. It consists of a ribonucleoprotein with a catalytic subunit with reverse transcriptase action (TERT) and an RNA fragment of sequence complementary to telomeric DNA (Terc: telomerase RNA component). Associated with the complex is dyskerin, a protein that contributes to its formation and stability. The catalytic site of telomerase adds deoxyribonucleotides using the RNA molecule as a template that hybridizes with the telomeric DNA strand (3′-OH end of the G strand). Through this mechanism, the enzyme is able to lengthen the telomere using a translocation movement each time it incorporates the complete hexanucleotide sequence (
Figure 2). Once the elongation of the G strand has concluded, the replication of the C strand occurs by the conventional system of polymerases. In this way, the telomeric shortening product of the terminal replication problem is solved [
15,
16,
17].
Figure 2. Telomerase activity. Schematic representation of telomere replication by telomerase, as well as the catalytic subunit with reverse transcriptase action (TERT) and the subunit containing the RNA template for telomere replication (Terc). Once the template RNA hybridizes with the telomeric DNA sequence of the G chain, at the 3′-OH end, the telomere polymerization process begins. Once the hexanucleotide sequence is added, the enzyme performs a translocation movement, and telomere elongation continues.
TERT is highly expressed in cells with high replication and self-renewal capacity, such as ESCs, germline cells, and most cancer cells. However, in adult stem cells (ASCs) and in activated lymphocytes, the activity of this enzyme was also demonstrated, although with much lower expression levels than in embryonic cells [
5,
18]. Except ASCs and activated lymphocytes, in the other somatic cells, telomerase activity decreases over time after birth, and subsequently, telomeres are shortened with cell divisions [
19]. Therefore, telomeric shortening limits the capacity for replicative expansion in telomerase-negative cells, constituting a barrier to achieving cellular immortality and functioning as a biological clock [
20]. On the other hand, there is evidence that even in somatic stem cells, telomerase levels are not satisfactory in avoiding telomeric shortening in the course of cellular aging [
21].
After 50 or 60 cell cycles, telomeric shortening leads to replicative senescence due to genomic instability associated with fusion events and chromosomal breakage. However, to avoid genetic chaos, some cells can overcome this phase by acquiring mutations in p53 and Rb genes and other genes, encoding for proteins linked to cell cycle control mechanisms and telomerase activation. A small part of the cell population acquires immortality through this pathway and proceeds to carcinogenesis [
20,
22].
Studies in mice highlighted that the defects in telomeres, and following telomerase re-activation, they are able to trigger the induction of malignant tumors [
23]. On the other hand, Barthel and colleagues observed the expression of TERT was present in almost 75% of the analyzed tumor samples, with 31% of samples exhibiting alterations in the TERT promoter and 53% showing methylation patterns [
24]. Aberrant expression of TERT is due to mutually exclusive mutations in the TERT promoter (TPM) (−57 A > C; −124 C > T; −138/−139 CC > TT; −146 C > T) that produce transcription factor binding sites of the ETS (E26 transformation-specific) family, such as GA binding protein (GABP) [
24,
25]. These mutations are mostly heterozygous and induce the allele-specific re-expression of TERT; recruiting GABP promotes an epigenetic change at the chromatin level, passing to its active form [
26].
TPMs are the most common non-coding driver of cancer mutations [
27]. Indeed, approximately 85% of skin tumors carry them [
28]. Likewise, they are coupled with a high expression of TERT and with poorer survival rates in several tumors, such as glioblastoma [
29] and meningioma [
30]. Furthermore, tumors that carry TPM shave a high risk of recurrence [
31]. Similarly, it is worth noting the existence of other pathways able to promote TERT expression during tumor, such as amplifying the TERT gene, hypermethylation of the promoter, and chromosomal rearrangement [
24,
32].
Moreover, the induction of the molecular mechanisms of DNA repair is also an important factor determining the cellular sequela in response to telomere changes. In this condition, NHEJ activation triggers chromosomal fusion, while the induction of the HDR mechanism could mediate the telomere lengthening through the ALT pathway [
20]. Consequently, activation of the telomere repair mechanisms could impact the progression of genomic instability and cancer induction by triggering the end’s union or obtaining cellular immortality through the ALT pathway. Because chromosome fusions and breakage of fused chromosomes could participate in cancer induction, further studies on DNA repair mechanisms that induce chromosomal fusions are required to better understand the role of telomeric shortening in cancer initiation [
20,
22].
2. Alternative Telomere Lengthening (ALT)
Even when telomerase is essential for obtaining cellular immortality, 10–15% of cancer cells can maintain stable telomere length through the ALT pathway [
6]. The ALT mechanism is common in sarcomas and tumors of the central and peripheral nervous system but rare in common cancers such as breast cancer, colon cancer, and lung cancer and absent in lymphoma and thymoma [
33]. Among cancers that arise in mesenchymal tissues, it was shown that 47% of osteosarcomas and 35% of soft tissue sarcomas (STS) use this mechanism of telomeric lengthening [
34]. Furthermore, several differences between and within STS have been described [
35]. In liposarcomas (LPS), ALT-positive tumors frequencies from 0% for well-differentiated LPS (characterized by amplification of the 12q13-15) to 80% for pleomorphic LPS, which show elevated levels of complex genomic alterations [
36].
Some evidence suggests that the ALT mechanism could be present in normal somatic cells. This was seen early in studies in mice [
37] and furthermore in primary human cells where the mechanism based on homologous recombination that resembles ALT seemed to be activated, transiently, in response to oxidative stress [
38] or X-ray damage [
39]. Similarly, intra-tumor heterogeneity was found in terms of telomere maintenance mechanisms (TMM) in neuroblastomas [
40] and osteosarcomas [
41], where several telomerase-positive cells have been detected, together with ALT-phenotyped cells.
In pluripotent stem cells (PSCs), telomere maintenance is carried out through telomerase expression, although the ALT pathway was found to play a fundamental role [
42,
43]. On the other hand, telomeres and the regulation of their length show marked differences between cancer cells and PSCs that use this mechanism. For example, tumor genomes employing the ALT pathway are unstable, exhibiting heterogeneous and dysfunctional telomeres, whereas PSCs possess long telomeres with a stable genome. The underlying mechanisms are not yet clear, but the ALT pathway is activated in PSCs by changes in epigenetic reprogramming [
43].
3. Coexistence of Telomerase and the ALT Pathway
The fusion of cell lines that express one of the two mechanisms allowed for obtaining hybrids where once the genome was stabilized, only one of the mechanisms remained active. Therefore, hybridized cells show only one telomere maintenance mechanism (TMM) by the expression of telomerase or by the activation of the ALT pathway [
60,
61,
62]. However, the ectopic overexpression of telomerase in ALT cells results in obtaining cell lines where both mechanisms coexist. In these cells, telomerase lengthens the shorter telomeres, while APBs and T-SCE can be detected with possible involvement in the maintenance of another subset of telomeres [
60,
63]. Therefore, the co-existence of both TMMs is only observed in the same cell when telomerase is over-expressed. Meanwhile, it has been shown that they can co-exist within the same tumor in different cells [
18]. The coexistence of ALT-positive cells and telomerase has been reported in several types of tumors, which supports the concept of intra-tumor heterogeneity. Indeed, TMM activation is not necessary for initial tumorigenesis. Still, it may occur during tumor expansion, involving the activation of both ALT and telomerase in different types of cells from the same tumor. Conversely, all early-phase tumor cells could have activated one of the two TMMs, with the possibility of changing this choice later [
64].