Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The RUNX family of transcription factors, including RUNX1, RUNX2, and RUNX3, are key regulators of development and can function as either tumor suppressors or oncogenes in cancer. Emerging evidence suggests that the dysregulation of RUNX genes can promote genomic instability in both leukemia and solid cancers by impairing DNA repair mechanisms. RUNX proteins control the cellular response to DNA damage by regulating the p53, Fanconi anemia, and oxidative stress repair pathways through transcriptional or non-transcriptional mechanisms.
- RUNX1
- RUNX2
- RUNX3
- DNA
- leukemia
1. Introduction
The RUNX family of proteins comprising RUNX1, RUNX2, and RUNX3 are master regulators of development [1][2]. As transcription factors, RUNX proteins bind to the consensus (Py)G(Py)GGT(Py) sequence on DNA through the evolutionarily conserved 128-amino acid RUNT domain. CBF-beta (CBFβ or core-binding factor β) is a critical dimerization partner that allosterically enhances the DNA-binding activity of RUNX factors during transcription [3][4]. However, RUNX proteins also perform transcription-independent roles in cells; they have been shown to interact with a growing list of central developmental regulators, epigenetic enzymes, and DNA repair factors in a cell and context-dependent manner [5]. Such combinatorial protein–protein interactions allow RUNX factors to function as tunable regulators of cell growth, differentiation, and carcinogenesis [1].
2. RUNX1 Leukemic Fusions in Hematopoietic Malignancies and Genomic Instability
RUNX1 is recurrently involved in chromosomal translocations in hematological malignancies, with almost 70 such chimeric fusions uncovered to date. The t(8;21)(q22;q22) karyotypic abnormality encoding RUNX1-RUNX1T1 (also known as RUNX1-ETO or AML-ETO) and t(12;21)(p13;q22) encoding ETV6-RUNX1 (also known as TEL-AML1) are among the most recurrent translocations in acute myeloid leukemia (AML) (10–20%) and B cell acute lymphoblastic leukemia (B-ALL) (25%), respectively [6][7]. The t(3;21) (q26;q22) translocation involving RUNX1 and MECOM1 (also known as EVI1) is frequently encountered in therapy-related myelodysplastic syndrome (MDS) and AML and during the blast crisis (BC) phase of chronic myeloid leukemia (CML) [8]. Inv16 (p13;q22), characterized by the CBFβ-MYH11 fusion, is a recurrent feature in AML (5–7%), in which RUNX function is impaired due to the inability of CBFβ to heterodimerize with RUNX [9]. Such neomorphic RUNX1 translocations mostly promote leukemogenesis by a novel gain-of-function or by dominantly inhibiting the function of the wild-type RUNX1 allele [10]. In the following section, researchers discuss evidence that RUNX1 leukemic fusions exacerbate genomic instability.
2.1. RUNX1-ETO and Genomic Instability
AML driven by RUNX1-ETO or AML1-ETO is a very well-studied AML subtype [11]. The RUNX1-ETO fusion gene has a structure comprising of the RUNX1 DNA-binding domain in addition to four conserved domains of the ETO protein, termed NHR1 to NHR4, that recruit transcriptional repressor complexes such as NCOR/HDAC/mSIN3a [12]. Since the RUNX1-ETO protein retains the DNA-binding domain of RUNX1 but lacks the carboxyl-terminal transactivation domain, the fusion binds to several RUNX1 target genes but represses their expression [13][14][15][16] and functions as a regulator of self-renewal and differentiation.
Notably, amongst the various RUNX1 leukemogenic fusions, the clearest mechanistic links between RUNX dysfunction and a “mutator” phenotype exist for t8;21 AML (Figure 1) [17]. However, RUNX1-ETO expression requires additional co-operating mutations in KIT, FLT3, RAS, ASXL1, and ZBTB7A, -9q, or –Y for the complete leukemic transformation of cells, and consistent with this idea, additional chromosomal aberrations are detected in almost 70% of t(8;21)-positive AML [18].
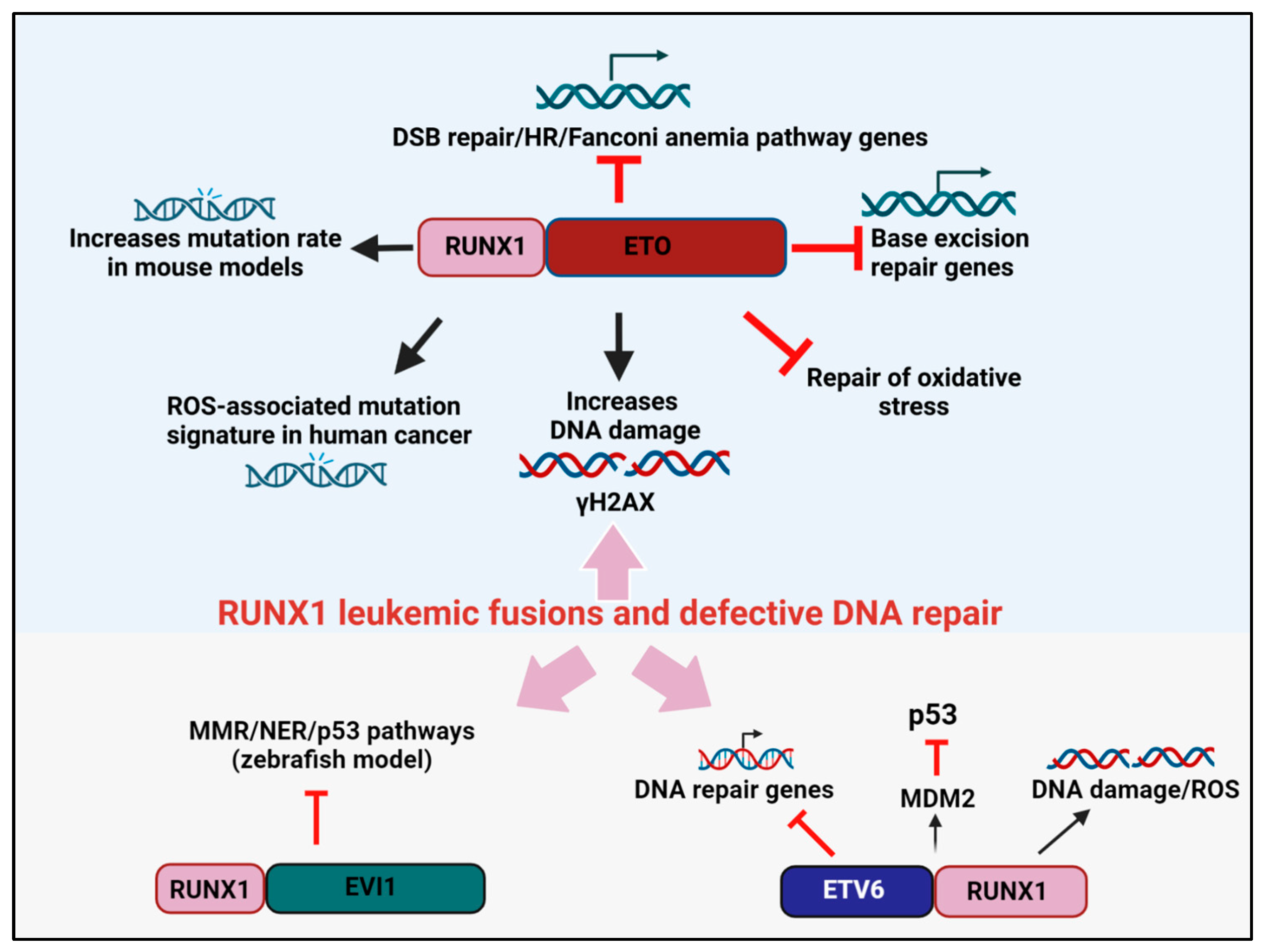
Figure 1. RUNX1 leukemic fusion proteins, DNA repair, and cancer. RUNX1-ETO attenuates the expression of genes involved in base excision repair, DSB repair, and the HR/FA pathways. RUNX1-ETO overexpression also reduces the efficiency of oxidative stress repair and induces the accumulation of γH2AX marked DSBs. In mouse models, RUNX1-ETO expression elevates mutation rates, while in human cancers, RUNX1-ETO expression induces a ROS-associated mutation signature. The leukemogenic fusion protein ETV6-RUNX1 blocks the expression of DNA repair genes and attenuates p53 signaling by increasing MDM2 expression. RUNX1-EVI1 was shown to reduce the expression of genes involved in MMR and NER in a zebrafish model.
Several studies have convincingly shown that RUNX1-ETO drives the acquisition of such co-operating mutations by downregulating the fidelity of DNA repair, thereby promoting a “mutator phenotype”. For instance, the overexpression of RUNX1-ETO reduced the expression of 17 DNA repair genes that participate in several DNA repair pathways, of which eight genes were involved in base excision repair (BER) (ADPRTL2, FEN1, OGG1, MPG, LIG3, POLB, POLD2, and POLD3) [19]. In assays that directly measure DNA repair, RUNX1-ETO-expressing cells were impaired in repairing oxidative lesions and had elevated levels of γH2AX, a marker of DNA double-strand breaks (DSBs) [19]. In an independent study by Krejci et al., RUNX1-ETO expression reduced the expression of genes from the ATM, ATR, and Fanconi anemia (FA) pathways of DNA repair [20]. Interestingly, HLTF, a protein that promotes replication fork reversal and limits multiple mechanisms of unrestrained DNA synthesis and replication stress, was also identified as a target of RUNX1 and was downregulated by RUNX1-ETO [21][22]. Likewise, Esposito et al. reported the downregulation of multiple FA and homologous recombination (HR) genes, including RAD51 and BRCA1/2, and the DSB sensor ATM, in RUNX-1 ETO-expressing leukemia [23].
In subsequent studies, to directly quantify the rate at which mutations are acquired, Forster et al. expressed RUNX1-ETO in the non-transformed TK6 lymphoblastoid cell line, and mutations at the PIG-A reporter gene was used as the read-out for genomic instability [24]. Remarkably, RUNX1-ETO expression was sufficient to predispose cells to elevated mutational acquisition both spontaneously and after exposure to genotoxic agents [24]. Likewise, in an in vivo model, RUNX1-ETO overexpression in a LacZ-plasmid (pUR288) expressing transgenic mouse resulted in an approximately 2-fold higher mutation rate over controls’ [20]. Independently, reactive oxygen species (ROS) have also emerged as a major etiology driving mutational accumulation in pediatric AML driven by RUNX1-ETO. Whole genome sequencing revealed that RUNX1-ETO-positive cases were associated with a higher prevalence of the ROS-associated SBS18 mutational signature, specifically owing to a high frequency of C>A transversions in this AML subtype. Additionally, it was shown that within RUNX1-ETO-fusion-positive AML cases, ROS-associated processes were not only contributing to mutations but also pro-oncogenic effects [25][26]. Together, the above studies highlight how the RUNX1-ETO oncoprotein exacerbates genomic instability through not one but multiple mechanisms, thereby permitting preleukemic cancer cells to acquire secondary hits which promote malignant progression.
2.2. ETV6-RUNX1 and Genomic Instability
The chimeric fusion protein ETV6-RUNX1 is a hallmark of B-ALL in which the N-terminus of the ETV6 gene is fused to almost the entire RUNX1 protein, and this event is thought to convert RUNX1 from a transcriptional activator to a repressor [27]. The early co-operating processes involved in the pathogenesis of this fusion protein have been difficult to determine. However, recent lineage tracing studies in mice have shown that the ETV6-RUNX1 clone is mostly preleukemic, and a second oncogenic hit appears essential for leukemic transformation [28].
A ETV6-RUNX1transgenic mouse model in which the fusion protein was expressed in precursor CD19+ B cells was examined for evidence of DNA damage accumulation and genomic instability [29]. Consistently, higher ROS and DNA damage accumulation were evident in the ETV6-RUNX1overexpressing mice, supporting the concept of higher mutability of the ETV6-RUNX1expressing genome (Figure 1). Independently, microarray comparisons between ETV6-RUNX1 knockdown and control ALL lines also revealed “DNA damage response” as a significant term. In this research, a total of 777 genes were substantially altered upon ETV6-RUNX1knockdown, and these comprised the DNA damage response genes (DRAM1, MDM2, CDKN1A, and PSD4) and genes regulated by p53, such as DDIT4 [30]. As further corroborating evidence, p53 signaling emerged as one of the central pathways deregulated in ETV6-RUNX1 expressing B-ALL compared to the fusion-negative B-ALL counterparts. Specifically, ETV6-RUNX1upregulated the transcription of MDM2, the negative regulator of p53; consistently blocking MDM2 through the inhibitor, nutlin, caused a surge in apoptosis [31]. Thus, ETV6-RUNX1-dependent p53 signaling impairment appears to be one of the driving forces underlying the development of a second oncogenic hit in this class of leukemia. In future work, a transgenic mouse model for ETV6-RUNX1generated in a p53-deficient background can clarify the precise contribution of p53 to ETV6-RUNX1-driven leukemogenesis.
2.3. RUNX1-EVI1 and Genomic Instability
The expression of RUNX1-EVI1 is common in therapy-induced MDS and during the BC transformation of chronic phase (CP) CML [32][33]. In this fusion protein, the N-terminal RUNT domain of RUNX1 is fused to almost the entire EVI1. In recent studies by Kellaway et al., RUNX1-EVI1 binding was shown to cause a redistribution of wild-type RUNX1 binding, which interfered with both the RUNX1 and EVI1 transcriptional programs [34]. While gene expression changes in DNA damage response genes were not reported in this research, a microarray analysis comparison of RUNX1-EVI1-driven transcriptional changes in a zebrafish model revealed the altered expression of mismatch repair (MMR) and nucleotide excision repair (NER) genes [35] (Figure 1). Moreover, given that RUNX1-EVI1 translocation is frequently retrieved after conventional chemotherapy, such as following hydroxyurea treatment in CML, it is tempting to speculate that this fusion gene provides a competitive advantage in the presence of DNA damage. Consistently, a proteomic analysis of the EVI1 protein-binding complexes unveiled EVI1 interaction with components of DNA repair and recombination [36]. One can speculate that RUNX1-EVI1 might create genomic instability in cancers by altering the interaction of EVI1 with DNA repair factors, although this model requires validation through rigorous biochemical studies.
3. RUNX3 Defects in Human Cancers and Genomic Instability
Unlike RUNX1, which is frequently mutated in human cancer, the RUNX3 gene is often transcriptionally silenced in cancer by CpG island DNA methylation or through EZH2-dependent H3K27me3 (histone H3 lysine 27 trimethylation) modification in the RUNX3 promoter [37]. RUNX3 can also be inactivated by cytoplasmic mis-localization and rarely through mutational inactivation (R122C mutation), as evident in gastric cancer [38][39]. DNA-damaging assaults such as smoking and ROS were shown to induce RUNX3 promoter hypermethylation [40][41]. Recently, in a novel mechanism of RUNX3 inactivation, Lee et al. showed hypoxia-induced methylation of RUNX3 by the enzyme G9a, and the methylated RUNX3, in turn, was attenuated in transactivation [42][43]. In this research, RUNX3 protein methylation was correlated with increased proliferation and initiation of tumorigenesis. Interestingly, Helicobacter pylori (H. pylori) infection itself has been shown to trigger RUNX3 inactivation through gradual step-wise promoter hypermethylation and accompanying silencing of the gene. Since H. pylori infection downregulates the expression of several DNA repair genes [44][45], it can be speculated that some of these transcriptional changes may be related to RUNX3 silencing, a model that needs to be experimentally tested in future work. Overall, based on the above observations, it can be hypothesized that RUNX3 silencing upon DNA damage might relieve a key anticancer barrier in epithelial tissues. Consistently, RUNX3 methylation was proposed as a ‘clock’ to determine the rate of bladder cancer progression [46].
Intriguingly, RUNX3 functions as an oncogene in NKT cell lymphoma, osteosarcoma, and ovarian cancers, mainly by increasing the transcription of MYC. By creating a transgenic mouse model, Douchi et al. showed that mutant RUNX3 R122C protein promotes gastric hyperplasia, and the upregulation of MYC was noted [39]. Thus, MYC upregulation is a common theme that is emerging when the downstream consequences of oncogenic RUNX3 expression are being examined. Given that MYC activation is associated with DNA replication stress and DNA damage [47], RUNX3 might activate DNA damage in these models through the transcriptional regulation of MYC (Figure 2).
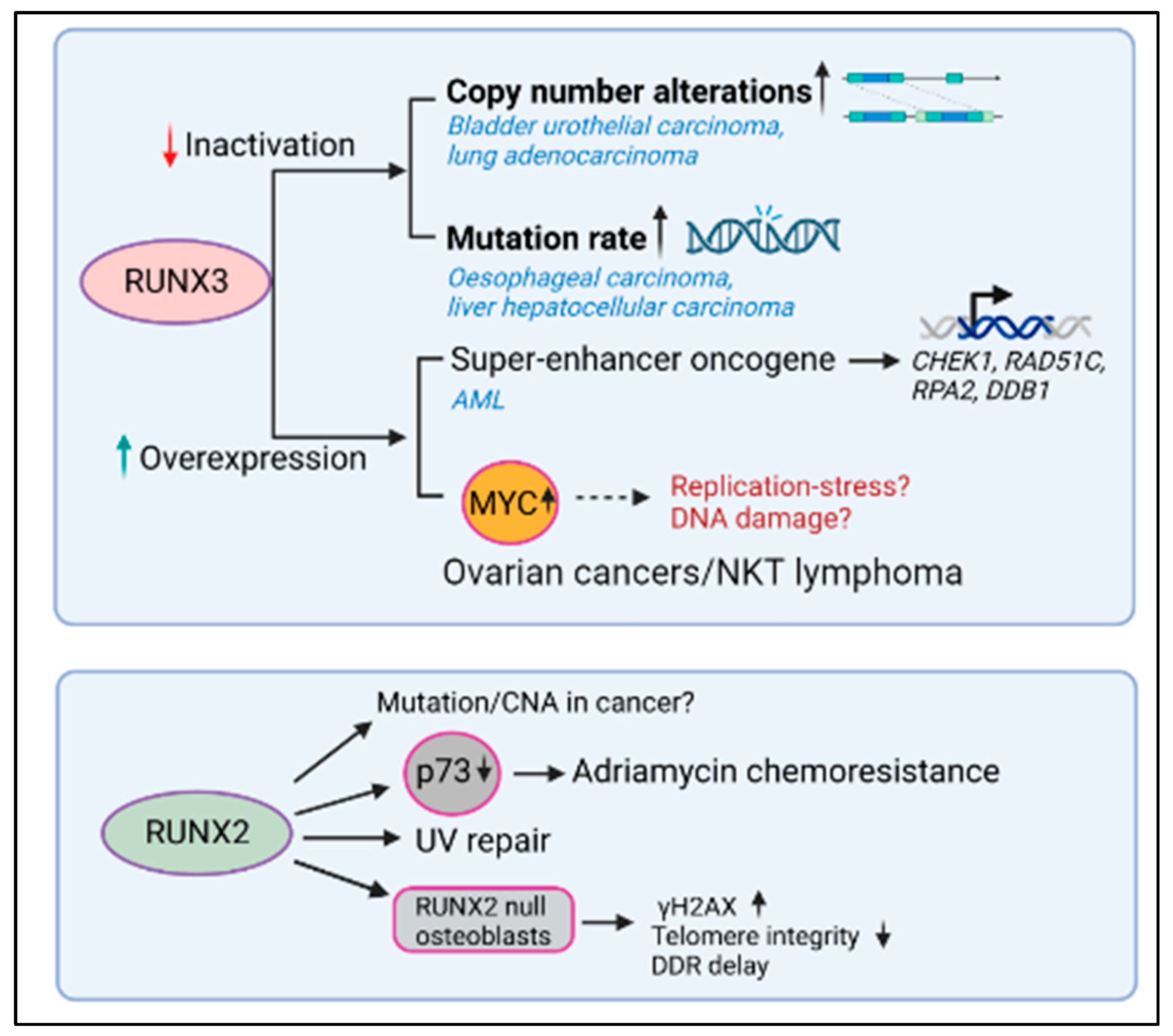
Figure 2. RUNX3 and RUNX3 dysregulation, DNA repair, and cancer. Lower RUNX3 transcript levels correlated with higher copy number alterations (CNAs) in bladder, urothelial carcinoma, and lung adenocarcinoma and with high mutation rate in esophageal carcinoma and liver hepatocellular carcinoma. In contrast, the higher levels of RUNX3 correlated with reduced DNA repair gene expression in AML and with higher levels of MYC in ovarian cancer and NKT cell lymphoma. While the expression of RUNX2 promoted the repair of UV-induced DNA damage and induced greater chemoresistance to adriamycin, the relationship between RUNX2 levels and mutational accumulation in the context of human malignancy remains unknown.
3.1. RUNX3 Inactivation and Genomic Instability
To examine how RUNX3 expression levels are related to genomic instability in human cancer, Tay et al. conducted a comprehensive genome-wide analysis of TCGA (The Cancer Genome Atlas) datasets [48]. Correlation coefficients between RUNX3 transcript levels and copy number alterations (CNA) or mutation counts were computed for all cancers. This analysis revealed that CNAs negatively correlated with RUNX3 expression most significantly in bladder urothelial carcinoma (BLCA) (n = 404, p = 9.28 × 10−6) and lung adenocarcinomas (LUAD) (n = 513, p = 8.78 × 10−7) (Figure 3). On the other hand, mutation rate negatively correlated with RUNX3 expression most significantly in esophageal carcinoma (ESCA) (n = 184, p = 4.3 × 10−6) and liver hepatocellular carcinoma (LIHC) (n = 366, p = 8.4 × 10−5). It can be argued that RUNX3 suppresses genomic instability more often in cancers having an etiological link to DNA damage, such as lung, bladder, and esophageal cancers which are predisposed by smoking, alcohol, and interstrand crosslinking agents, respectively. It can be hypothesized that in such cancers, lower RUNX3 probably lowers proapoptotic signaling by p53, allowing cells to survive in the presence of DNA damage (see below, RUNX and p53 crosstalk).
3.2. RUNX3 Activation and Genomic Instability
In contrast, RUNX3 was identified as a super-enhancer-associated oncogene in AML and was one of the most highly expressed genes in this cancer type. As a transcription factor, RUNX3 is bound to the promoter of cell cycle-related genes in both normal and AML cells. However, within AML cells, RUNX3 is also bound to the promoters of DNA repair genes (CHEK1, RAD51C, RPA2, and DDB1), antiapoptotic genes (BCL2, BCL2L10, BCL2L12, and MCL1), and genes implicated in leukemogenesis (MYC, CD93, KIT, IKZF2, FTO, and SOX4) [49] (Figure 3). In this research, RUNX3 knockdown inhibited leukemic progression by inducing DNA damage and higher apoptosis. Thus, as an oncogene, RUNX3 overexpression induces a higher resistance to DNA damage-induced apoptosis. It can be inferred from the above studies that RUNX3 levels are critical in determining whether DNA damage signals enter the apoptotic pathway via p53. The influence of RUNX proteins on p53 signaling strengths will be discussed in the following sections.
4. RUNX2 Defects in Cancers and Genomic Instability
In contrast to RUNX1 and RUNX3, which may function as oncogenes or tumor suppressors, RUNX2 is mostly overexpressed and oncogenic in human cancer. RUNX2 has promigratory effects on breast, prostate, and thyroid cancer cells, osteosarcoma, and melanoma cells and has emerged as a key regulator of cancer metastasis. RUNX2 overexpression increases the expression of genes involved in invasion and metastasis, such as MMP9, MMP13, OPN, VEGF, and IL-8, epithelial-mesenchymal transition factors such as SNAI2, SMAD3, and SOX9, and motility genes such as FAK/PTK2 and TNL1. RUNX2 also promotes metastasis by activating the AKT/PI3K, YAP-TAZ, TGFβ, and WNT signaling pathways and angiogenesis, thereby driving positive feedback loops that advance cancer progression [50].
RUNX2 Dysregulation and Genomic Instability
In one of the earliest studies on the relationship between RUNX proteins and DNA repair, primary RUNX2-null osteoblasts were shown to accumulate spontaneous γH2AX foci, experience loss of telomere integrity, and have delayed DNA damage response [51]. Subsequently, RUNX2 was shown to form functional complexes with BAZ1B, RUVBL2, and INTS3 and influences UV repair by complexing with H2AX and decreasing histone H3 lysine 9 acetylation levels [52]. More recently, RUNX2 was shown to promote the phosphorylation of H2AX at 142, thus favoring apoptosis instead of repair [53]. In this research, RUNX2 recruitment to osteogenic target genes was dependent on DNA damage and led to an enhancement in calcification during aging and chronic disease. In the context of malignancy, overexpressed RUNX2 regulates chemosensitivity by attenuating the transcriptional activity and proapoptotic function of p73 after exposure to the chemotherapeutic adriamycin, supporting an oncogenic role for RUNX2 in chemoresistance [54]. However, it remains unknown if RUNX2 overexpression is causally relatedly to genomic instability and mutational accumulation in human cancers.
This entry is adapted from the peer-reviewed paper 10.3390/cells12081106
References
- Ito, Y.; Bae, S.-C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95.
- Mevel, R.; Draper, J.E.; Lie-A-Ling, M.; Kouskoff, V.; Lacaud, G. RUNX transcription factors: Orchestrators of development. Development 2019, 146, dev148296.
- Tahirov, T.H.; Inoue-Bungo, T.; Morii, H.; Fujikawa, A.; Sasaki, M.; Kimura, K.; Shiina, M.; Sato, K.; Kumasaka, T.; Yamamoto, M.; et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell 2001, 104, 755–767.
- Ito, Y. RUNX Genes in Development and Cancer: Regulation of Viral Gene Expression and the Discovery of RUNX Family Genes. Adv. Cancer Res. 2008, 99, 33–76.
- Chuang, L.S.H.; Ito, K.; Ito, Y. RUNX family: Regulation and diversification of roles through interacting proteins. Int. J. Cancer 2013, 132, 1260–1271.
- Peterson, L.F.; Zhang, D.-E. The 8;21 translocation in leukemogenesis. Oncogene 2004, 23, 4255–4262.
- Zelent, A.; Greaves, M.; Enver, T. Role of the TEL-AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukaemia. Oncogene 2004, 23, 4275–4283.
- Rubin, C.M.; Larson, R.; Anastasi, J.; Winter, J.N.; Thangavelu, M.; Vardiman, J.W.; Rowley, J.D.; Le Beau, M.M. t(3;21)(q26;q22): A recurring chromosomal abnormality in therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood 1990, 76, 2594–2598.
- Liu, P.; Tarle, S.A.; Hajra, A.; Claxton, D.F.; Marlton, P.; Freedman, M.; Siciliano, M.J.; Collins, F.S. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993, 261, 1041–1044.
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082.
- Lam, K.; Zhang, D.E. RUNX1 and RUNX1-ETO: Roles in hematopoiesis and leukemogenesis. Front. Biosci. 2012, 17, 1120–1139.
- Sun, X.-J.; Wang, Z.; Wang, L.; Jiang, Y.; Kost, N.; Soong, T.D.; Chen, W.-Y.; Tang, Z.; Nakadai, T.; Elemento, O.; et al. A stable transcription factor complex nucleated by oligomeric AML1–ETO controls leukaemogenesis. Nature 2013, 500, 93–97.
- Ptasinska, A.; Assi, S.A.; Martinez-Soria, N.; Imperato, M.R.; Piper, J.; Cauchy, P.; Pickin, A.; James, S.R.; Hoogenkamp, M.; Williamson, D.; et al. Identification of a Dynamic Core Transcriptional Network in t(8;21) AML that Regulates Differentiation Block and Self-Renewal. Cell Rep. 2014, 8, 1974–1988.
- Stengel, K.R.; Ellis, J.D.; Spielman, C.L.; Bomber, M.L.; Hiebert, S.W. Definition of a small core transcriptional circuit regulated by AML1-ETO. Mol. Cell 2020, 81, 530–545.e5.
- Thiel, V.N.; Giaimo, B.D.; Schwarz, P.; Soller, K.; Vas, V.; Bartkuhn, M.; Blätte, T.J.; Döhner, K.; Bullinger, L.; Borggrefe, L.; et al. Heterodimerization of AML1/ETO with CBFbeta is required for leukemogenesis but not for myeloproliferation. Leukemia 2017, 31, 2491–2502.
- Rejeski, K.; Duque-Afonso, J.; Lubbert, M. AML1/ETO and its function as a regulator of gene transcription via epigenetic mechanisms. Oncogene 2021, 40, 5665–5676.
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434.
- Mrózek, K.; Bloomfield, C.D. Clinical Significance of the Most Common Chromosome Translocations in Adult Acute Myeloid Leukemia. JNCI Monogr. 2008, 2008, 52–57.
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761.
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.-S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199.
- Cheng, C.K.; Chan, N.P.H.; Wan, T.S.K.; Lam, L.Y.; Cheung, C.H.Y.; Wong, T.H.Y.; Ip, R.K.L.; Wong, R.S.M.; Ng, M.H.L. Helicase-like transcription factor is a RUNX1 target whose downregulation promotes genomic instability and correlates with complex cytogenetic features in acute myeloid leukemia. Haematologica 2016, 101, 448–457.
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7.
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W.E. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490.
- Forster, V.J.; Nahari, M.H.; Martinez-Soria, N.; Bradburn, A.K.; Ptasinska, A.; Assi, S.A.; Fordham, S.E.; McNeil, H.; Bonifer, C.; Heidenreich, O.; et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia 2015, 30, 251–254.
- Mandell, J.D.; Fisk, J.N.; Cyrenne, E.; Xu, M.L.; Cannataro, V.L.; Townsend, J.P. Not only mutations but also tumorigenesis can be substantially attributed to DNA damage from reactive oxygen species in RUNX1::RUNX1T1-fusion-positive acute myeloid leukemia. Leukemia 2022, 36, 2931–2933.
- Gunnarsson, R.; Yang, M.; Olsson-Arvidsson, L.; Biloglav, A.; Behrendtz, M.; Castor, A.; Paulsson, K.; Johansson, B. Single base substitution mutational signatures in pediatric acute myeloid leukemia based on whole genome sequencing. Leukemia 2021, 35, 1485–1489.
- Romana, S.P.; Mauchauffé, M.; Le Coniat, M.; Chumakov, I.; Le Paslier, D.; Berger, R.; Bernard, O. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood 1995, 85, 3662–3670.
- Rodríguez-Hernández, G.; Casado-García, A.; Isidro-Hernández, M.; Picard, D.; Raboso-Gallego, J.; Alemán-Arteaga, S.; Orfao, A.; Blanco, O.; Riesco, S.; Prieto-Matos, P.; et al. The Second Oncogenic Hit Determines the Cell Fate of ETV6-RUNX1 Positive Leukemia. Front. Cell Dev. Biol. 2021, 9, 704591.
- Kantner, H.P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia 2013, 15, 1292–1300.
- Fuka, G.; Kauer, M.; Kofler, R.; Haas, O.A.; Panzer-Grumayer, R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS ONE 2011, 6, e26348.
- Kaindl, U.; Morak, M.; Portsmouth, C.; Mecklenbräuker, A.; Kauer, M.; Zeginigg, M.; Attarbaschi, A.; Haas, O.A.; R Panzer-Grümayer, R. Blocking ETV6/RUNX1-induced MDM2 overexpression by Nutlin-3 reactivates p53 signaling in childhood leukemia. Leukemia 2014, 28, 600–608.
- Nucifora, G.; Rowley, J. The AML1 Gene in the 8;21 and 3;21 Translocations in Chronic and Acute Myeloid Leukemia. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 595–605.
- Yin, C.C.; Cortes, J.; Barkoh, B.; Hayes, K.; Kantarjian, H.; Jones, D. t(3;21)(q26;q22) in myeloid leukemia: An aggressive syndrome of blast transformation associated with hydroxyurea or antimetabolite therapy. Cancer 2006, 106, 1730–1738.
- Kellaway, S.G.; Keane, P.; Kennett, E.; Bonifer, C. RUNX1-EVI1 disrupts lineage determination and the cell cycle by interfering with RUNX1 and EVI1 driven gene regulatory networks. Haematologica 2020, 106, 1569–1580.
- Shen, L.; Zhu, J.; Chen, F.; Lin, W.; Cai, J.; Zhong, J. RUNX1-Evi-1 fusion gene inhibited differentiation and apoptosis in myelopoiesis: An in vivo study. BMC Cancer 2015, 15, 970.
- Bard-Chapeau, E.A.; Gunaratne, J.; Kumar, P.; Chua, B.Q.; Muller, J.; Bard, F.A.; Blackstock, W.; Copeland, N.G.; Jenkins, N.A. EVI1 oncoprotein interacts with a large and complex network of proteins and integrates signals through protein phosphorylation. Proc. Natl. Acad. Sci. USA 2013, 110, E2885–E2894.
- Fujii, S.; Ito, K.; Ito, Y.; Ochiai, A. Enhancer of Zeste Homologue 2 (EZH2) Down-regulates RUNX3 by Increasing Histone H3 Methylation. J. Biol. Chem. 2008, 283, 17324–17332.
- Goh, Y.-M.; Cinghu, S.; Hong, E.T.H.; Lee, Y.-S.; Kim, J.-H.; Jang, J.-W.; Li, Y.-H.; Chi, X.-Z.; Lee, K.-S.; Wee, H.; et al. Src Kinase Phosphorylates RUNX3 at Tyrosine Residues and Localizes the Protein in the Cytoplasm. J. Biol. Chem. 2010, 285, 10122–10129.
- Douchi, D.; Yamamura, A.; Matsuo, J.; Lee, J.-W.; Nuttonmanit, N.; Lim, Y.H.M.; Suda, K.; Shimura, M.; Chen, S.; Pang, S.; et al. A Point Mutation R122C in RUNX3 Promotes the Expansion of Isthmus Stem Cells and Inhibits Their Differentiation in the Stomach. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1317–1345.
- Chen, L.-M.; Nergard, J.C.; Ni, L.; Rosser, C.J.; Chai, K.X. Long-Term Exposure to Cigarette Smoke Extract Induces Hypomethylation at the RUNX3 and IGF2-H19 Loci in Immortalized Human Urothelial Cells. PLoS ONE 2013, 8, e65513.
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic changes induced by oxidative stress in colorectal cancer cells: Methylation of tumor suppressor RUNX3. Tumor Biol. 2012, 33, 403–412.
- Lee, S.H.; Kim, J.; Kim, W.-H.; Lee, Y.M. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 2008, 28, 184–194.
- Lee, S.H.; Hyeon, D.Y.; Yoon, S.-H.; Jeong, J.-H.; Han, S.-M.; Jang, J.-W.; Nguyen, M.P.; Chi, X.-Z.; An, S.; Hyun, K.-G.; et al. RUNX3 methylation drives hypoxia-induced cell proliferation and antiapoptosis in early tumorigenesis. Cell Death Differ. 2020, 28, 1251–1269.
- Kolinjivadi, A.M.; Sankar, H.; Choudhary, R.; Tay, L.S.; Tan, T.Z.; Murata-Kamiya, N.; Voon, D.C.-C.; Kappei, D.; Hatakeyama, M.; Krishnan, V.; et al. The H. pylori CagA Oncoprotein Induces DNA Double Strand Breaks through Fanconi Anemia Pathway Downregulation and Replication Fork Collapse. Int. J. Mol. Sci. 2022, 23, 1661.
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep. 2015, 11, 1703–1713.
- Wolff, E.M.; Liang, G.; Cortez, C.C.; Tsai, Y.C.; Castelao, J.E.; Cortessis, V.K.; Tsao-Wei, D.D.; Groshen, S.; Jones, P.A. RUNX3 Methylation Reveals that Bladder Tumors Are Older in Patients with a History of Smoking. Cancer Res 2008, 68, 6208–6214.
- Curti, L.; Campaner, S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. Int. J. Mol. Sci. 2021, 22, 6168.
- Tay, L.S.; Krishnan, V.; Sankar, H.; Chong, Y.L.; Chuang, L.S.H.; Tan, T.Z.; Kolinjivadi, A.M.; Kappei, D.; Ito, Y. RUNX Poly(ADP-Ribosyl)ation and BLM Interaction Facilitate the Fanconi Anemia Pathway of DNA Repair. Cell Rep. 2018, 24, 1747–1755.
- Zhang, W.; Ma, Q.; Long, B.; Sun, Z.; Liu, L.; Lin, D.; Zhao, M. Runt-Related Transcription Factor 3 Promotes Acute Myeloid Leukemia Progression. Front. Oncol. 2021, 11, 4065.
- Cohen-Solal, K.A.; Boregowda, R.K.; Lasfar, A. RUNX2 and the PI3K/AKT axis reciprocal activation as a driving force for tumor progression. Mol. Cancer 2015, 14, 137.
- Zaidi, S.K.; Pande, S.; Pratap, J.; Gaur, T.; Grigoriu, S.; Ali, S.A.; Stein, J.L.; Lian, J.B.; van Wijnen, A.J.; Stein, G.S. Runx2 deficiency and defective subnuclear targeting bypass senescence to promote immortalization and tumorigenic potential. Proc. Natl. Acad. Sci. USA 2007, 104, 19861–19866.
- Yang, S.; Quaresma, A.J.; Nickerson, J.A.; Green, K.M.; Shaffer, S.A.; Imbalzano, A.N.; Martin-Buley, L.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; et al. Subnuclear domain proteins in cancer cells support the functions of RUNX2 in the DNA damage response. J. Cell Sci. 2015, 128, 728–740.
- Cobb, A.M.; Yusoff, S.; Hayward, R.; Ahmad, S.; Sun, M.; Verhulst, A.; D’Haese, P.C.; Shanahan, C.M. Runx2 (Runt-Related Transcription Factor 2) Links the DNA Damage Response to Osteogenic Reprogramming and Apoptosis of Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2021, 41, 1339–1357.
- Ozaki, T.; Sugimoto, H.; Nakamura, M.; Hiraoka, K.; Yoda, H.; Sang, M.; Fujiwara, K.; Nagase, H. Runt-related transcription factor 2 attenuates the transcriptional activity as well as DNA damage-mediated induction of pro-apoptotic TAp73 to regulate chemosensitivity. FEBS J. 2014, 282, 114–128.
This entry is offline, you can click here to edit this entry!