Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Circulating glucocorticoids increase during stress. Chronic stress, characterized by a sustained increase in serum levels of cortisol, has been associated in different cases with an increased risk of cancer and a worse prognosis. Glucocorticoids can promote gluconeogenesis, mobilization of amino acids, fat breakdown, and impair the body's immune response. Therefore, conditions that may favor cancer growth and the acquisition of radio- and chemo-resistance. We found that glucocorticoid receptor knockdown diminishes the antioxidant protection of murine B16-F10 (highly metastatic) melanoma cells, thus leading to a drastic decrease in their survival during interaction with the vascular endothelium. The BRAFV600E mutation is the most commonly observed in melanoma patients. Recent studies revealed that VMF/PLX40-32 (vemurafenib, a selective inhibitor of mutant BRAFV600E) increases mitochondrial respiration and reactive oxygen species (ROS) production in BRAFV600E human melanoma cell lines. Early-stage cancer cells lacking Nrf2 generate high ROS levels and exhibit a senescence-like growth arrest. Thus, it is likely that a glucocorticoid receptor antagonist (RU486) could increase the efficacy of BRAF-related therapy in BRAFV600E-mutated melanoma. In fact, during early progression of skin melanoma metastases, RU486 and VMF induced metastases regression. However, treatment at an advanced stage of growth found resistance to RU486 and VMF. This resistance was mechanistically linked to overexpression of proteins of the Bcl-2 family (Bcl-xL and Mcl-1 in different human models). Moreover, melanoma resistance was decreased if AKT and NF-κB signaling pathways were blocked. These findings highlight mechanisms by which metastatic melanoma cells adapt to survive and could help in the development of most effective therapeutic strategies.
- melanoma
- stress
- glucocorticoids
- Nrf2
- antioxidant defenses
- cancer
1. Glucocorticoids, Nrf2 and the Antioxidant Defense of Melanoma Cells
The transcription activator Nrf2 is the master regulator of the antioxidant response and upregulates the expression of antioxidant and detoxifying enzymes [100]. Nrf2 has a protective role in UV-induced oxidative stress, DNA damage, and apoptosis of melanocytes [101]. Given the important contribution of UV radiation for ROS formation, it is not surprising that Nrf2 activity is induced by UV in melanocytes [100]. Nrf1 and Nrf2 transcription factors, upon activation by oxidative stress, form heterodimers with different factors, i.e., Maf and Jun, to bind to the antioxidant/electrophile response element (ARE/EpRE) and regulate the transcription of oxidative stress/cytoprotection-related genes [59,102]. This is important because oncogene (i.e., KRAS, BRAF or MYC)-induced Nrf2 transcription activity associates to increases in melanoma growth and pharmacological resistance [103,104,105]. Elevated Nrf2 expression and a high GSH/GSSG ratio in melanoma are correlated with a deeper Breslow index, invasive/metastatic phenotype, and poor survival [106,107,108]. In that sense, it was shown that GSH protects melanoma from oxidative stress, contributing to its survival [107,109]. These results are in agreement with Beberok et al. who demonstrated that treatment with the antibiotic Lomefloxacin induce oxidative stress, GSH depletion and apoptosis in the COLO829 melanoma cell line [110]. Moreover, N-acetylcysteine (a classical mucolytic drug) can promote melanoma metastases spread, a fact suggesting that caution should be taken when administering GSH promoters to cancer patients [111]. Furthermore, the link between Nrf2 and immune tolerance has already been shown in lung adenocarcinoma, where Keap1 mutations are present in up to 20% and lead to permanent Nrf2 activation [112]. This is an important question in the case of metastatic melanomas where the anti-PD-1 immune therapy represent the only optional treatment [113,114].
Since glucocorticoids increase ROS generation in metastatic B16-F10 melanoma cells [98] and also in breast cancer cells [115], we investigated if the decrease in antioxidant enzyme activities in invasive B16-F10 cells (iB16) knockdown for the GR (iB16-shGR) was associated with changes in nuclear Nrf1 and/or Nrf2. Nuclear Nrf2, and not Nrf1, decreased in iB16-shGR cells isolated from lung or liver metastatic foci compared to control iB16 cells [116]. This is a fact that may be key since increased Nrf2 transcription activity has been correlated with aggressiveness in different human cancers [117,118,119]. However, other authors have found just opposite results postulating that GR signaling represses the antioxidant response, e.g., by inhibiting the histone acetylation mediated by Nrf2 [120], or by forming a glucocorticoid-GR complex that migrates to the nucleus where it binds to glucocorticoid response elements and ARE/EpRE sequences [121,122]. These controversial results should be analyzed in the light of the actual doses of glucocorticoids administered. Ligand-occupied GR induces or represses the transcription of thousands of genes by direct binding to DNA response elements and/or by physically associating with other transcription factors, thus involving a vast array of molecular interactions [123]. It is then essential to differentiate between pathophysiological and pharmacological levels, the latter being much higher (as reported in [120,122]) and potentially causing very different results. Indeed, biological stressors can positively or negatively affect antioxidant enzymes depending on the time and levels of exposure [124]. In fact, exposure to physiological stressors induces the production of ROS and oxidative stress in, e.g., the rat liver [125]. More importantly, a meta-analysis of glucocorticoids as modulators of oxidative stress concluded that glucocorticoids promote oxidative stress [126], which cannot be the result of improving the antioxidant defenses. Although, glucocorticoids are currently used against different cancers [127], they can also induce cancer resistance, a still unclear effect that may promote growth and metastases [127,128]. In fact, at pathophysiological levels, glucocorticoid signaling is antiapoptotic in cells of epithelial origin and in many malignant solid tumors subjected to cytotoxic therapy [89,129,130,131]. It was shown, in the immortalized human mammary epithelial cell line MCF10A, that GR-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1 [132]. In agreement with these ideas, GR antagonism has been shown to promote apoptosis in solid tumor cells [133].
Importantly, at high levels or long-term exposure of ROS, p53 expression (promoted by DNA damage) increases, activating prooxidant genes, interfering with the Nrf2-dependent transcription of ARE/EpRE-containing promoters (and, thereby, inhibiting the Nrf2-mediated survival response), and potentially resulting in cell death [134]. However, particularly in highly aggressive human cancers, the p53 protein is reduced, lost, or mutated [135]. In this scenario, we used AS101 (ammonium tri-chloro(dioxoethylene-O,O′-)tellurate), a synthetic compound which has immunomodulating properties [136] and increases expression of wild-type p53 [137]. We observed that AS101-induced up-regulation of p53 in iB16 melanoma cells caused a decrease in antioxidant enzyme expression without affecting the nuclear levels of Nrf2 [116]. An effect that was reversed by using anti-p53 antisense oligonucleotides [116]. Thus, proving that p53 can suppress the Nrf2-dependent transcription of antioxidant enzymes in metastatic melanoma cells. Interestingly GR activation may lead to inhibition of p53-induced apoptosis, an effect observed in, e.g., MCF-10Amyc cells [138]. In agreement with this concept, in estrogen receptor-positive breast cancer cells, low GR expression has been linked to higher p53 expression [139]. Indeed, p53 can form a complex with the glucocorticoid that causes a cytoplasmic sequestration of both molecular structures [140]. These facts suggest a close link among GRs, p53 and Nrf2 which could be involved in regulating growth and spread of BRAFV600E-mutated melanoma cells. Figure 1 summarizes, as a working hypothesis, potential molecular interactions that may involve GRs and the Nrf2-dependent antioxidant defenses in melanoma cells.
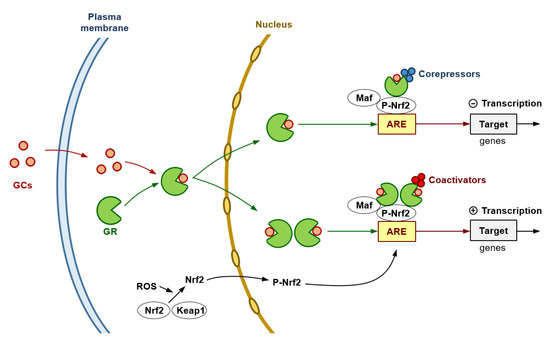
Figure 1. Glucocorticoids and the Antioxidant Defense of Melanoma Cells. The GR is encoded by the NRC31 gene which can produce a number of receptor isoforms, the GRα being the primary receptor involved in glucocorticoid (GC) signaling. The cytosolic GR complexes with different proteins, i.e., Hsp90, Hsp70 and the FK506-binding protein 4. GCs diffuse through the plasma membrane into the cytoplasm and binds to the GR resulting in the release of heat shock proteins. Based on the two-part model proposed by Gerber et al. [142], the cytoplasmic GR interacts with glucocorticoids, thus causing a conformational change and nuclear translocation. GR interacts with both the DNA and other transcriptional machinery to orchestrate its genomic effects through three main mechanisms: direct binding to glucocorticoid response elements, transcription factor tethering, and binding of composite elements within the DNA [143]. Hypothetically, at high pharmacological levels of GCs, primary repression could result from an excessive amount of GR monomers tethering to the ARE-Nrf2 complex (Nrf2 dimerizes with a basic region-leucine zipper bZIP protein and binds to the ARE to activate gene transcription), then leading to recruitment of corepressors. In this mechanism, GR associates with NF-κB or AP-1 [144,145], thus resulting in repression of their activity in a process attributed to recruitment of transcriptional corepressors such as the nuclear receptor co-repressor 1 and the histone deacetylase 2 [146]. At lower extracellular levels of GCs (pathophysiological levels), GR homodimers (predominantly formed) would recruit coactivators (such as the steroid receptor coactivator-1, SRC-1; or the GR-interacting protein-1, GRIP-1) [147] and induce the transcription of genes encoding antioxidant/defense enzyme activities. This double model may help to reconcile the controversy in the results obtained by different groups.
Upon interaction of circulating melanoma cells with the vascular endothelium, a cascade of molecular events associates to the classical docking and rolling, i.e., attachment to the endothelial cells, release of proinflammatory cytokines, ROS and RNS, and tumor cytotoxicity [141]. Independently of the tumor location (liver, lung, or subcutaneous), in metastatic melanoma cells, GR knockdown decreased the expression and activities of γ-GCS, superoxide dismutase 1 and 2, catalase, glutathione peroxidase, and glutathione reductase, inducing a reduction in GSH levels [111]. Facts showing that GR knockdown compromises the antioxidant defense of melanoma cells, and increases the endothelium-induced tumor cytotoxicity [116]. Hence limiting their invasive capacity. This opens up potential therapeutic application in case selective GR blockers may show pharmacological efficacy under in vivo conditions.
2. Combined Glucocorticoid Receptor Antagonism and BRAF Inhibition Promotes Regression of Early Melanoma Metastases
Mifepristone (RU486) is a steroidal antiprogestogen (IC50 = 0.025 nM), as well as an antiglucocorticoid (IC50 = 2.2 nM), and antiandrogen (IC50 = 10 nM) to a much lesser extent [148]. Its relative binding affinity at the GR is more than three times that of dexamethasone and more than ten times that of cortisol [149]. The proposed mechanism of action of RU486 it is a competitive binding to the GR that prevents the dissociation of the heat shock proteins from the receptor avoiding its subsequent translocation to the nucleus and transcriptional activity [150]. RU486 does not bind to the estrogen receptor or the mineralocorticoid receptor [151]. Research work has revealed that progesterone can inhibit human melanoma cell growth. The mechanism of inhibition is due to autophagy and this effect of progesterone is not mediated through progesterone receptor [152]. Down-modulation or pharmacological inhibition of androgen receptors suppresses melanomagenesis, with increased intratumoral infiltration of macrophages and, in an immune-competent mouse model, cytotoxic T cells [153]. However, intracellular signaling derived from activation of progesterone or androgen receptors is different from that derived from GRs [154]. Recent studies have demonstrated cytotoxic and anti-metastatic effects of RU486 in vitro and in clinical trials involving meningioma, colon, breast, and ovarian cancers (e.g., Ritch et al. [155]), whereas Alvarez et al. [156] demonstrated that RU486 impedes the proliferation of uveal melanoma cells (a highly metastatic and drug resistant cancer). Furthermore, metapristone (the most active metabolite of RU486) inhibited cell viability and induced early and late apoptosis in B16-F10 and A375 melanoma cells [157]. Metapristone treatment resulted in decreased of Akt and ERK phosphorylation and of Bcl-2 and facilitated overexpression of p53 and Bax in A375 cells. In addition, metapristone suppressed cell migration and invasion by down-regulating the expression of matrix metalloproteinases (2 and 9), N-cadherin and vimentin, while E-cadherin expression was up-regulated [157].
The BRAFV600E mutation is the most commonly observed in patients, represents more than 90% of BRAF mutations in melanoma, and can be detected early during melanoma development [158]. B-Raf signaling create a balance between a pro-oncogenic signal and a senescent proliferative arrest. Interestingly, in human fibroblasts BRAFV600E-induced senescence was bypassed by the addition of glucocorticoids (albeit at pharmacological doses), which allowed their cancer transformation [159].
Vemurafenib (VMF)/PLX40-32 (a selective inhibitor of mutant BRAFV600E) was the first molecularly targeted therapy to be licensed in the US and Europe for treatment of advanced melanoma. Its mechanism of action involves selective inhibition of the mutated BRAF V600E kinase that leads to reduced signaling through the aberrant mitogen-activated protein kinase pathway [160]. It has been reported that VMF increases mitochondrial respiration-linked ROS generation in BRAFV600E melanoma cell lines [161]. However, VMF also induces HO-1 upregulation in primary BRAFV600E melanoma cell lines, limiting the efficacy of the drug and reducing the cancer cell recognition and killing by natural killer cells [162]. Thus, possibly, a GR antagonist could increase the efficacy of BRAF-related therapy in BRAFV600E-mutated melanoma. To test this hypothesis, we studied the effect of RU486 [163] on the antioxidant defense of different human BRAFV600E melanoma cell lines. We found that in vivo administration of RU486 to mice bearing metastatic BRAFV600E-mutated melanoma cells decreases Nrf2- and redox state-related enzyme activities and, in parallel, increases ROS production [109]. Further experiments showed that combined treatment with RU486 and VMF strongly inhibits BRAFV600E-mutated metastatic melanoma growth in vivo [109]. Importantly, melanoma growth inhibition was only observed if RU486 and VMF were administered simultaneously. However, if administration of VMF was delayed, the inhibitory effect of the association practically disappeared [109]. Thus, suggesting that, despite RU486 administration, melanoma cells can spontaneously develop anti-VMF resistance. Indeed, it is well known that the anti-melanoma effects of VMF are sort-lived, and that patients present tumor relapse in a short period after treatment [164,165]. In fact, melanoma cells showing acquired resistance to VMF have high rates of mitochondrial respiration associated with elevated mitochondrial oxidative stress [161]. Thus, suggesting that targeting the antioxidant defense could be the right therapeutic choice.
It is also worth to mention that the most common adverse effects of VMF treatment, i.e., pyrexia, arthralgia or skin rash, are usually treated with dexamethasone [166,167]. However, based on the above discussion, this therapy should be reconsidered as it has been recently recommended by the Oncological Endocrinology research group of the Italian Society of Endocrinology [168].
3. Anti-Death Adaptations Related to the Bcl-2 Family of Proteins in Advanced BRAFV600E-Mutated Melanoma Metastases
Recent research indicate that a stress-like state promotes overexpression of fos, hsp70 and ubb, all required for adaptation to diverse cellular stresses. This state has a higher tumor seeding capabilities compared to non-stressed cells, and confers intrinsic resistance to MEK inhibitors, commonly used in melanoma treatment [169]. Furthermore, this stress-like program can be induced by, e.g., heat shock, and promotes resistance to both MEK and BRAF inhibitors in human melanomas [169]. Further mechanisms of acquired melanoma resistance involve activation of the MAPK pathway. The PI3K-PTEN-AKT pathway is a 2nd resistance pathway, which often overlaps with the MAPK pathway [170,171]. Acquired resistance to MAPK pathway targeted therapies (BRAF/MEK inhibitors) develops in most patients at approx. 12 months [172]. Interestingly, it was also shown that GR-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival [173]. Moreover, the MEK/ERK signaling pathway regulates expression of different Bcl-2-related proteins and survival in, e.g., human pancreatic cancer cells [174,175]. We observed that different melanoma cells, surviving after RU486 treatment, down regulated expression of different Bcl-2-related pro-death genes (i.e., bax, bak, bid), whereas upregulated anti-death bcl-xl and mcl-1 [109]. Thus, we investigated if inhibition of Bcl-xL or Mcl-1 could improve the anti-melanoma effects of RU486 and VMF. Previously, dexamethasone was found to inhibit TRAIL-induced apoptosis of thyroid cancer cells via Bcl-xL induction [176]. Indeed, the treatment with RU486 + VMF + UMI-77 (UMI77 is a selective small-molecule inhibitor of Mcl-1 [177]) or RU486 + VMF + WEHI77 (WEHI77 is Bcl-xL-selective BH3 mimetic [178]) almost induced a complete regression of advanced BRAFV600E-mutated melanoma metastases depending on which of these Bcl-2-related proteins was preferentially overexpressed in the different human BRAFV600E melanomas tested [109]. These findings are very relevant since melanoma regression was also associated to an increase in host survival [109], and because BRAFV600E mutation can also be present in other malignant neoplasms such as hairy-cell leukemia, colon carcinoma, ovarian low-grade serous carcinoma, Langerhans cell histiocytosis and Erdheim-Chester disease, glial neoplasms and thyroid carcinoma [179].
This entry is adapted from the peer-reviewed paper 10.3390/cells12030418
This entry is offline, you can click here to edit this entry!