Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
TP53 mutations are less frequent in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) than in solid tumors, except in secondary and therapy-related MDS/AMLs, and in cases with complex monosomal karyotype. As in solid tumors, missense mutations predominate, with the same hotspot mutated codons (particularly codons 175, 248, 273). As TP53-mutated MDS/AMLs are generally associated with complex chromosomal abnormalities, it is not always clear when TP53 mutations occur in the pathophysiological process.
- mutant p53
- cancer
- myelodysplasia
- acute myeloid leukemia
1. Introduction
The transcription factor p53, encoded by the TP53 gene in humans, is one of the most studied tumor suppressor proteins. In normal cells, p53 activity is low, but in response to DNA damage and numerous other stress signals, p53 levels rise dramatically and result in the activation and transcription of genes with important roles in cell cycle arrest, senescence, apoptosis, metabolism, and differentiation [1]. The sum of these activities is to ensure that an abnormal cell fails to arise and proliferate. During normal hematopoiesis, p53 activities preserve genome integrity and regulate several cellular processes that maintain the normal stem cell pool and serve as a barrier to tumorigenesis [2][3].
Perturbations in p53 activity or p53-dependent pathways are required for the development of most cancers [4], and there is evidence in many situations to suggest that the restoration or reactivation of physiological p53 function may be therapeutic [5][6][7][8]. Various mechanisms are responsible for the disruption of p53 activity in cancer, mainly deletion or mutation of the TP53 gene, and overexpression of the p53 negative regulators Mdm2 and Mdm4 [9][10][11]. Several isoforms are encoded by the TP53 gene and appear to play different roles in tumorigenesis/cancer progression [12] or response to treatment, for example in acute myeloid leukemia (AML) [13]. Regardless of the mechanism behind p53 dysfunction, the downstream consequences are profound due to the very large spectrum of biological activities in which p53 is normally implicated. The clinical correlation between p53 mutational status in cancer cells and resistance to treatment has been studied since the 1990s. In breast cancer, presence of a TP53 mutation is associated with resistance to doxorubicin [14][15]. Similarly, ovarian cancer patients harboring a TP53 mutation are less sensitive to treatment with cisplatin [16][17]. Moreover, the association of TP53 mutation with chemoresistance and poor prognosis has also been observed in lung [18], gastric, and colorectal cancers [19], as well as in hematological malignancies [20][21]. Apart from triggering chemoresistance, p53 mutants are also able to attenuate cancer response to radiotherapy [14][19][22].
Myelodysplastic syndromes (MDS) are a clonal hematopoietic stem cell disorders characterized by cell dysplasia, ineffective hematopoiesis that leads to cytopenias (mainly anemia) and a variable risk of progression to AML [23]. Furthermore, AML is characterized by clonal expansion of undifferentiated myeloid precursors, resulting in impaired hematopoiesis and life-threatening cytopenias. Among the prognostic factors identified in both malignancies, presence of a mutation in the TP53 gene indicates a particularly dismal prognosis irrespective of the treatment administered [24].
2. Characteristics of TP53 Mutations in MDS/AML
TP53 gene mutations are reported in about 50% of solid tumors, but only 5–10% of de novo myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). However, while these mutations are rare in MDS/AML with a normal karyotype (1%), they are seen in 40–50% of secondary and therapy-related cases [20][25][26][27], and in 80% of complex monosomal karyotypes (CK) that include 17p and/or 5q deletion [28][29]. Other inactivating mechanisms of wild-type p53 function include overexpression of MDM2 and MDM4, negative regulators of p53, in 20–30% and 40–50% of AML cases, respectively [30][31][32][33][34]. Unfortunately, treatment with Mdm2 inhibitors has not so far demonstrated a substantial effect in these cases [35]. Inactivation of p14-ARF, a positive regulator of p53, has been more rarely reported in AML [32][36][37].
As in solid tumors, most TP53 mutations in MDS/AML cluster in exons 4 to 8 that encode the DNA binding domain [38][39]. Nearly 75% of the mutants are missense: recurrent hotspot variants are also observed, as in solid tumors, and at the same frequent codons such as the contact mutants at codons 248 and 273, and the structural mutants at codons 175 and 245 [40]. The number of cooperating driver mutations is usually very low in TP53-mutated MDS/AML, and even absent in 85% of the cases [41].
TP53 alteration in MDS/AML can be monoallelic or biallelic [42]. Monoallelic mutations, with point mutation of only one TP53 allele, represent 25 to 30% of the cases. They are particularly seen in MDS with isolated 5q deletion [43][44][45][46], but can also be discovered in various MDS and AML with non-complex karyotype by systematic NGS analysis, generally at low variant allelic frequency (VAF) [42]. In 70 to 75% of the cases, TP53 alterations are biallelic, generally resulting from point mutation on one TP53 allele (usually missense) and loss of the other allele, through 17p deletion or monosomy 17. The great majority of MDS/AMLs with biallelic alteration have indeed complex monosomal karyotype resulting in 17p deletion, very often with 5q deletion [47]. On the other hand, monoallelic TP53 mutations can lead to genomic instability, chromosomal loss and chromothripsis (also known as “chromosome shattering” that leads to large structural rearrangements in chromosomes [48]) including loss of heterozygosity (LOH) through 17p deletion [49], and therefore resulting in a biallelic hit. TP53 mutations, especially in the biallelic state, are associated with resistance to most treatments, including chemotherapy, hypomethylating agents, and even allogeneic stem cell transplantation, as the risk of relapse post-transplant is very high, at least in case of biallelic alterations [50][51][52][53]. Monoallelic mutations are also, to a lesser extent, associated with a certain resistance to treatment, for example to lenalidomide–an immuno-modulatory drug used in MDS with del(5q) [43][54], but have generally a more limited impact on survival.
3. When Do TP53 Mutations Arise in MDS and AML Cells?
Whether TP53 mutations occur early or late in the evolution of MDS and AML remains uncertain in most situations. In well-performed studies in therapy-related MDS/AML, it was found that cells harboring the leukemia-specific TP53 mutation preexisted to the first cancer and were positively selected by treatment for this cancer (chemotherapy or radiation) to which they were resistant [55][56]. Progressive evolution was seen through genomic instability and chromothripsis with inactivation of the second TP53 allele, as seen above [49] (Figure 1). TP53 mutations are also among the mutations found in healthy individuals with clonal hematopoiesis of indeterminate potential (CHIP), who have an increased risk of developing various blood cancers [57][58]. However, DNMT3A, TET2, and ASXL1 mutations largely predominate in CHIPs seen in patients with no prior history of treated cancer, and CHIPs with TP53 mutation are mainly seen in patients having received chemotherapy or radiotherapy for a prior cancer, where they increase the risk of therapy-related MDS/AML, potentially by the selection mechanism just described above [55][59][60][61][62][63][64].
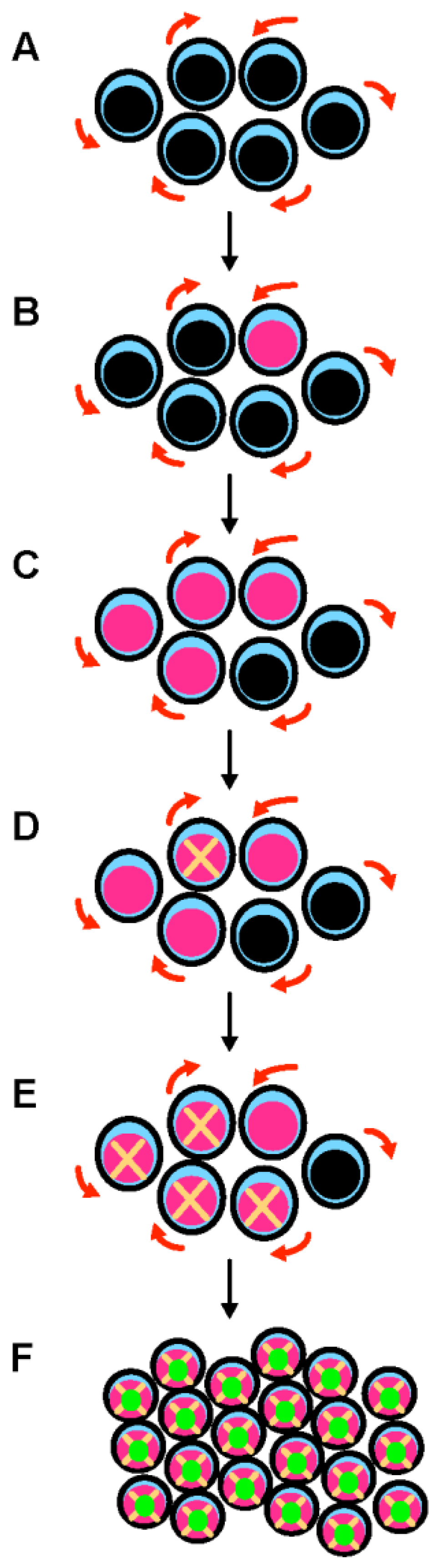
Figure 1. A multistep process leads to the development of TP53-mutated myelodysplastic syndrome and acute myeloid leukemia. (A) Hematopoietic stem cells (HSCs) are the only cells within the hematopoietic system that possess the potential for both multipotency (i.e., the potential to differentiate into all of the mature blood cell type) and self-renewal (i.e., the potential to make more stem cells, thus perpetuating the stem cell pool throughout life). Self-renewal is illustrated with red arrows. (B) A mutation in the TP53 gene can emerge in a hematopoietic stem cell (pink nucleus). This mutation can appear spontaneously and is selected in response to various stresses, including exposure to cytotoxic treatments. This mutation can be induced by these cytotoxic treatments (such as chemotherapy or radiation therapy) or by workplace exposure to toxic chemicals and carcinogenic substances such as benzene. (C) Mutant p53 confers a competitive advantage in the stem cell compartment. At this early stage, cells express both wild type and mutant p53 proteins (potential dominant-negative effect). (D) TP53-mutated HSCs acquire chromosomal changes and gene mutations that either result from mutant p53-related genomic instability and chromothripsis, or are induced by cytotoxic chemotherapy/radiation. (E) These acquired genetic changes further enhance the fitness of TP53-mutated HSCs. (F) Further chromosomal changes lead to the loss of the wild type TP53 allele by 17p deletion or monosomy 17. Loss of wild type TP53 functions and potential gain-of-function activities of mutant p53 are responsible for the development of overt acute myeloid leukemia.
It is still unclear in other MDS/AML situations when TP53 mutations arise. In MDS with isolated 5q deletion, detectable monoallelic TP53 mutations, often at very low VAF and sometimes multiple, are found in 20% of the patients at diagnosis [43][44][45][46]. Whether they occur during disease evolution or very early in minor undetectable clones (by conventional methods) that could undergo positive selection, including by treatment with lenalidomide, is unclear. In those MDS with isolated del(5q) and monoallelic TP53 mutation, as in the therapy-related model described above, progression to AML is generally preceded by acquisition of a complex karyotype, including del(17p) and biallelic TP53 inactivation [65][66] (Figure 2).
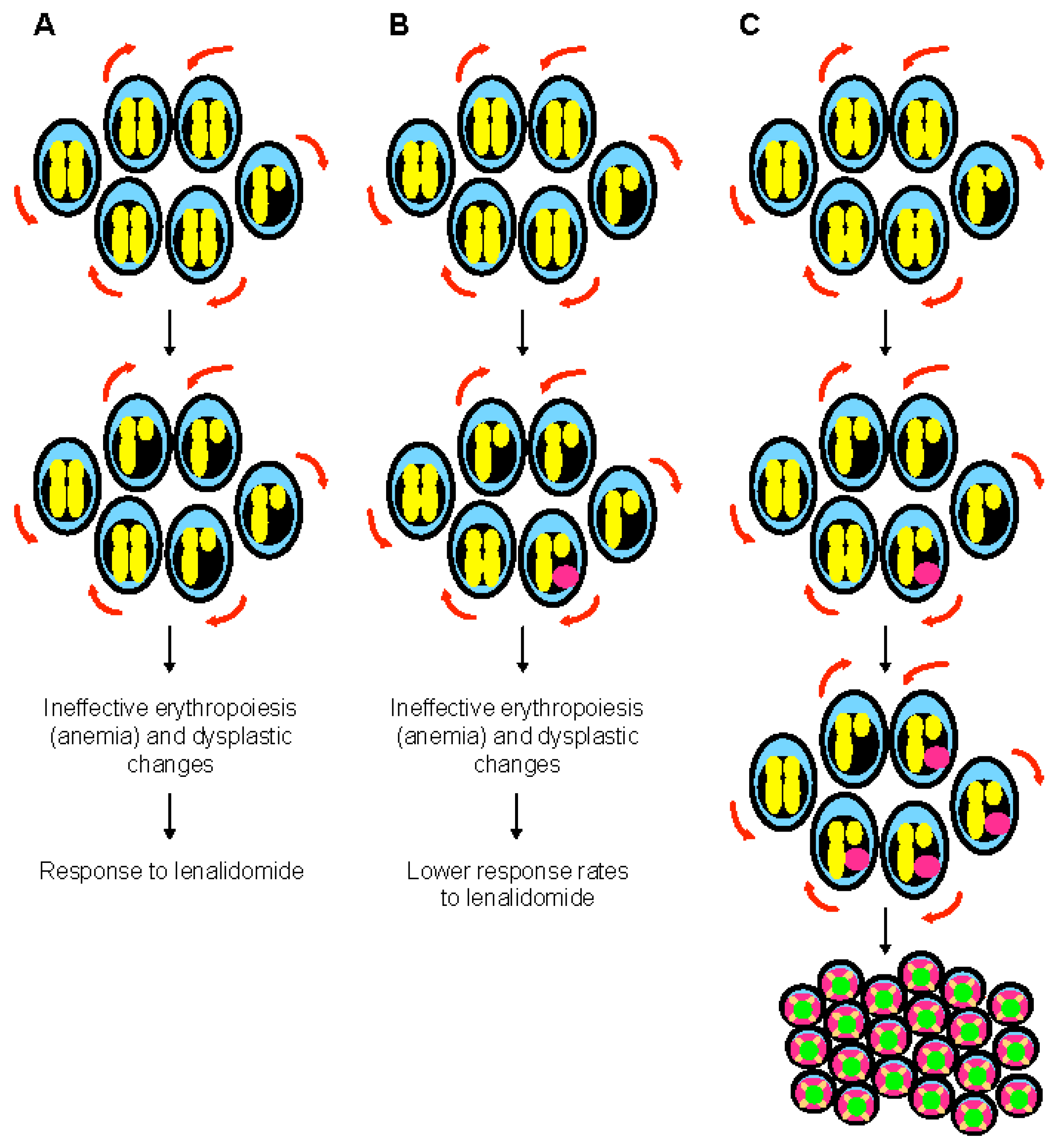
Figure 2. The special case of myelodysplastic syndrome with 5q deletion. (A) Deletion of 5q chromosome is somatically acquired and heterozygous. This chromosomal abnormality is present in the hematopoietic stem cell compartment and can be found in all lineages. The clinical phenotype of 5q- syndrome (i.e., ineffective erythropoiesis and dysplastic changes) is related to haploinsufficient gene expression of several genes such as RPS14, APC, and EGR1. Self-renewal is illustrated with red arrows. (B) Monoallelic TP53 mutations are seen at diagnosis in almost 20% of patients with 5q- syndrome, generally at low variant allelic frequency. These mutations are associated with resistance to lenalidomide, but have generally a limited impact on survival. At this stage, karyotype is non-complex. (C) Monoallelic TP53 mutations are associated with genomic instability and chromothripsis in myelodysplastic syndrome with isolated 5q deletion. Progression to higher risk myelodysplastic syndrome and acute myeloid leukemia is preceded by acquisition of a complex karyotype, including 17p deletion and biallelic TP53 inactivation.
In myeloproliferative neoplasms, TP53 mutations are seen in about 20% of the cases with progression to MDS/AML. They appear to be late events, although once again small TP53 mutant clones, undetectable by conventional techniques, may have occurred earlier in the disease course [67][68][69][70][71][72][73]. When TP53-mutated clones appear during the disease course would be important to determine, as their early detection in myeloid malignancies could allow to target them pharmacologically, potentially preventing progression to full-blown biallelic TP53 MDS/AML.
TP53 mutations could represent early leukemogenic events in many situations, and they have been indeed reported in pre-leukemic hematopoietic stem cells (HSCs) of AML patients [74][75]. Major roles of TP53 are related to cell-cycle control, DNA repair and apoptosis. Dysregulation of these pivotal functions might be an alternative mechanism to epigenetic modifications in establishing a proleukemogenic state in HSCs [76]. By transforming HSCs into pre-leukemic stem cells (pre-LSCs), TP53 mutations substantially contribute to the development of AML and its resistance to conventional treatments [55][77][78]. Moreover, TP53-mutated pre-LSCs retain their ability to differentiate into mature blood cells both in patient-derived mouse xenografts and patients with AML [78]. Clonogenic assays revealed that patient-specific TP53 mutations are present in the vast majority of HSC-derived colonies (median, 97%; range, 45 to 100%), with only a paucity of cooperating mutations in known cancer genes, as seen above. Copy-number alterations, as already described, appear to be secondary events after the onset of a first TP53 mutation [79][80].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11041152
References
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Pant, V.; Quintás-Cardama, A.; Lozano, G. The p53 pathway in hematopoiesis: Lessons from mouse models, implications for humans. Blood 2012, 120, 5118–5127.
- Aloni-Grinstein, R.; Shetzer, Y.; Kaufman, T.; Rotter, V. p53: The barrier to cancer stem cell formation. FEBS Lett. 2014, 588, 2580–2589.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288.
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Brown Swigart, L.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571.
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575.
- Wang, Y.; Suh, Y.-A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintás-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923.
- Wasylishen, A.R.; Lozano, G. Attenuating the p53 Pathway in Human Cancers: Many Means to the Same End. Cold Spring Harb. Perspect. Med. 2016, 6, a026211.
- Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288.
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545.
- Bergh, J.; Norberg, T.; Sjögren, S.; Lindgren, A.; Holmberg, L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995, 1, 1029–1034.
- Aas, T.; Børresen, A.L.; Geisler, S.; Smith-Sørensen, B.; Johnsen, H.; Varhaug, J.E.; Akslen, L.A.; Lønning, P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814.
- Shelling, A.N. Role of p53 in drug resistance in ovarian cancer. Lancet 1997, 349, 744–745.
- Petty, R.; Evans, A.; Duncan, I.; Kurbacher, C.; Cree, I. Drug resistance in ovarian cancer—The role of p53. Pathol. Oncol. Res. 1998, 4, 97–102.
- Horio, Y.; Takahashi, T.; Kuroishi, T.; Hibi, K.; Suyama, M.; Niimi, T.; Shimokata, K.; Yamakawa, K.; Nakamura, Y.; Ueda, R. Prognostic significance of p53 mutations and 3p deletions in primary resected non-small cell lung cancer. Cancer Res. 1993, 53, 1–4.
- Hamada, M.; Fujiwara, T.; Hizuta, A.; Gochi, A.; Naomoto, Y.; Takakura, N.; Takahashi, K.; Roth, J.A.; Tanaka, N.; Orita, K. The p53 gene is a potent determinant of chemosensitivity and radiosensitivity in gastric and colorectal cancers. J. Cancer Res. Clin. Oncol. 1996, 122, 360–365.
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994, 84, 3148–3157.
- Wilson, W.H.; Teruya-Feldstein, J.; Fest, T.; Harris, C.; Steinberg, S.M.; Jaffe, E.S.; Raffeld, M. Relationship of p53, bcl-2, and tumor proliferation to clinical drug resistance in non-Hodgkin’s lymphomas. Blood 1997, 89, 601–609.
- Koch, W.M.; Brennan, J.A.; Zahurak, M.; Goodman, S.N.; Westra, W.H.; Schwab, D.; Yoo, G.H.; Lee, D.J.; Forastiere, A.A.; Sidransky, D. p53 mutation and locoregional treatment failure in head and neck squamous cell carcinoma. J. Natl. Cancer Inst. 1996, 88, 1580–1586.
- Fenaux, P.; Platzbecker, U.; Ades, L. How we manage adults with myelodysplastic syndrome. Br. J. Haematol. 2020, 189, 1016–1027.
- Papaemmanuil, E.; Döhner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901.
- Slingerland, J.M.; Minden, M.D.; Benchimol, S. Mutation of the p53 gene in human acute myelogenous leukemia. Blood 1991, 77, 1500–1507.
- Fenaux, P.; Jonveaux, P.; Quiquandon, I.; Laï, J.L.; Pignon, J.M.; Loucheux-Lefebvre, M.H.; Bauters, F.; Berger, R.; Kerckaert, J.P. P53 gene mutations in acute myeloid leukemia with 17p monosomy. Blood 1991, 78, 1652–1657.
- Fenaux, P.; Preudhomme, C.; Quiquandon, I.; Jonveaux, P.; Laï, J.L.; Vanrumbeke, M.; Loucheux-Lefebvre, M.H.; Bauters, F.; Berger, R.; Kerckaert, J.P. Mutations of the P53 gene in acute myeloid leukaemia. Br. J. Haematol. 1992, 80, 178–183.
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Peng, J.; Tang, G.; Goswami, M.; Young, K.H.; Singh, R.; Medeiros, L.J.; et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J. Hematol. Oncol. 2015, 8, 45.
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 3, 1747–1758.
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305.
- Bueso-Ramos, C.E.; Yang, Y.; deLeon, E.; McCown, P.; Stass, S.A.; Albitar, M. The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993, 82, 2617–2623.
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982.
- Dey, A.; Tergaonkar, V.; Lane, D.P. Double-edged swords as cancer therapeutics: Simultaneously targeting p53 and NF-kappaB pathways. Nat. Rev. Drug Discov. 2008, 7, 1031–1040.
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96.
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). OncoTargets Ther. 2019, 12, 2903–2910.
- Müller-Tidow, C.; Metzelder, S.K.; Buerger, H.; Packeisen, J.; Ganser, A.; Heil, G.; Kügler, K.; Adigüzel, G.; Schwäble, J.; Steffen, B.; et al. Expression of the p14ARF tumor suppressor predicts survival in acute myeloid leukemia. Leukemia 2004, 18, 720–726.
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Mol. Cell 2013, 51, 46–56.
- Lai, J.L.; Preudhomme, C.; Zandecki, M.; Flactif, M.; Vanrumbeke, M.; Lepelley, P.; Wattel, E.; Fenaux, P. Myelodysplastic syndromes and acute myeloid leukemia with 17p deletion. An entity characterized by specific dysgranulopoïesis and a high incidence of P53 mutations. Leukemia 1995, 9, 370–381.
- Soenen, V.; Preudhomme, C.; Roumier, C.; Daudignon, A.; Laï, J.L.; Fenaux, P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood 1998, 91, 1008–1015.
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30.
- Prochazka, K.T.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Pabst, G.; Wölfler, A.; Zebisch, A.; Berghold, A.; Döhner, K.; Sill, H. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica 2019, 104, 516–523.
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556.
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011, 29, 1971–1979.
- Sebaa, A.; Ades, L.; Baran-Marzack, F.; Mozziconacci, M.-J.; Penther, D.; Dobbelstein, S.; Stamatoullas, A.; Récher, C.; Prebet, T.; Moulessehoul, S.; et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes Chromosomes Cancer 2012, 51, 1086–1092.
- Scharenberg, C.; Giai, V.; Pellagatti, A.; Saft, L.; Dimitriou, M.; Jansson, M.; Jädersten, M.; Grandien, A.; Douagi, I.; Neuberg, D.S.; et al. Progression in patients with low- and intermediate-1-risk del(5q) myelodysplastic syndromes is predicted by a limited subset of mutations. Haematologica 2017, 102, 498–508.
- Lodé, L.; Ménard, A.; Flet, L.; Richebourg, S.; Loirat, M.; Eveillard, M.; Le Bris, Y.; Godon, C.; Theisen, O.; Gagez, A.-L.; et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 2018, 103, e143–e146.
- Volkert, S.; Kohlmann, A.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer 2014, 53, 402–410.
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670.
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20.
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698.
- Bally, C.; Adès, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.-J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 2014, 38, 751–755.
- Desoutter, J.; Gay, J.; Berthon, C.; Ades, L.; Gruson, B.; Geffroy, S.; Plantier, I.; Marceau, A.; Helevaut, N.; Fernandes, J.; et al. Molecular prognostic factors in acute myeloid leukemia receiving first-line therapy with azacitidine. Leukemia 2016, 30, 1416–1418.
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036.
- Martinez-Høyer, S.; Deng, Y.; Parker, J.; Jiang, J.; Mo, A.; Docking, T.R.; Gharaee, N.; Li, J.; Umlandt, P.; Fuller, M.; et al. Loss of lenalidomide-induced megakaryocytic differentiation leads to therapy resistance in del(5q) myelodysplastic syndrome. Nat. Cell Biol. 2020, 22, 526–533.
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555.
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547.
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4.
- Wong, T.N.; Miller, C.A.; Jotte, M.R.M.; Bagegni, N.; Baty, J.D.; Schmidt, A.P.; Cashen, A.F.; Duncavage, E.J.; Helton, N.M.; Fiala, M.; et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat. Commun. 2018, 9, 455.
- Chen, S.; Gao, R.; Yao, C.; Kobayashi, M.; Liu, S.Z.; Yoder, M.C.; Broxmeyer, H.; Kapur, R.; Boswell, H.S.; Mayo, L.D.; et al. Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia 2018, 32, 850–854.
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649.
- Chen, S.; Liu, Y. p53 involvement in clonal hematopoiesis of indeterminate potential. Curr. Opin. Hematol. 2019, 26, 235–240.
- Miller, P.G.; Steensma, D.P. Implications of Clonal Hematopoiesis for Precision Oncology. JCO Precis. Oncol. 2020, 4, 639–646.
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541.
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 2009, 23, 203–206.
- Stoddart, A.; Fernald, A.A.; Wang, J.; Davis, E.M.; Karrison, T.; Anastasi, J.; Le Beau, M.M. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood 2014, 123, 1069–1078.
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712.
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490.
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410.
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Gotlib, J.R.; Schrijver, I.; Zehnder, J.L.; Arber, D.A. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod. Pathol. 2015, 28, 706–714.
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adélaïde, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2017, 102, e11–e14.
- Kubesova, B.; Pavlova, S.; Malcikova, J.; Kabathova, J.; Radova, L.; Tom, N.; Tichy, B.; Plevova, K.; Kantorova, B.; Fiedorova, K.; et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: Association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia 2018, 32, 450–461.
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333.
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553.
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317.
- Heuser, M. Therapy-related myeloid neoplasms: Does knowing the origin help to guide treatment? Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 24–32.
- Lal, R.; Lind, K.; Heitzer, E.; Ulz, P.; Aubell, K.; Kashofer, K.; Middeke, J.M.; Thiede, C.; Schulz, E.; Rosenberger, A.; et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood 2017, 129, 2587–2591.
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71.
- Gualberto, A.; Aldape, K.; Kozakiewicz, K.; Tlsty, T.D. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 5166–5171.
This entry is offline, you can click here to edit this entry!