PARK7/DJ-1 is expressed in almost all, if not all, human cells [
83]; it is primarily localized in the cytoplasm, however, its occurrence in the mitochondria [
84] and nucleus [
85] and also its secretion into the extracellular space was reported [
86]. Previously, the expression of PARK7/DJ-1 was mainly investigated regarding malignant tumors and neurodegenerative diseases. In malignant diseases, the increased expression of PARK7/DJ-1 was demonstrated in glioblastoma, non-small cell lung, thyroid, breast, hepatocellular, and colorectal carcinoma [
87]. In addition, the elevated PARK7/DJ-1 expression was closely correlated with the poor survival of patients with colorectal and pancreas cancers [
87,
88,
89]. On the contrary, the reduced expression and/or the increased amount of dysfunctional overoxidized form of PARK7/DJ-1 was found in the brain of patients with various neurodegenerative disorders, including PD, AD, Huntington’s disease, and ischemic stroke [
90].
4. Role of PARK7/dj-1 in the Pathogenesis of Neurodegenerative Diseases
The connection between PARK7/DJ-1 and neurodegenerative diseases was first suggested nearly two decades ago when it was identified as a causative factor in rare inherited forms of PD [
7]. Since then, the role of PARK7/DJ-1 in neurodegenerative diseases has been intensively studied, and it has become clear that PARK7/DJ-1 may play a role not only in PD but in almost all neurological diseases associated with oxidative stress, inflammation, and tissue damage [
9,
126,
127].
4.1. Genetic Evidence for the Role of PARK7/DJ-1 in Parkinson’s Disease
The group of Parkinson’s disease molecules involves 23 genes located on different chromosomes and having different functions [
128]. However, one thing that is common in the members of the PARK family is that they are all associated with the higher risk of PD [
129]. This relationship was first suggested in 2003 when Bonifati and colleagues found a large (about 14 kb) deletion and a missense mutation (Leucine166Proline, L166P) in the PARK7/DJ-1 gene in a Dutch and an Italian family, leading to the identification of PARK7/DJ-1 as a causative gene for familial PD with recessive inheritance [
7]. Since then, more than 20 PARK7/DJ-1 mutations have been associated with PD [
130].
4.2. Role of PARK7/DJ-1 in Parkinson’s Disease
Experimental data suggest that PARK7/DJ-1 plays a protective role in neurodegenerative diseases via its antioxidant properties. Indeed, the lack of PARK7/DJ-1 in stem cell-derived neurons and SH-SY5Y cells resulted in increased vulnerability to oxidative stress [
131,
132,
133]. In addition, the neuroprotective effect of the administration of recombinant PARK7/DJ-1 was demonstrated in the rodent model of 6-hydroxydopamine (6-OHDA) and MG-132 treatment-induced PD [
134]. Furthermore, it has been demonstrated that pharmacological protection of PARK7/DJ-1 against overoxidation preserves its antioxidant properties. Indeed, Miyazaki et al. and Kitamura et al. identified small molecule compounds, including UCP0054277, UCP0054278, and Compound 23 (Comp23), that can bind to the C106 region of PARK7/DJ-1 and keep it in reduced, biologically active form [
8,
135]. The protective effects of these compounds against oxidative stress were confirmed in hydrogen peroxide- (H
2O
2) treated wild type and PARK7/DJ-1-knockdown SH-SY5Y neuronal cells [
8,
135,
136]. In further experiments, they also demonstrated that administration of UCP0054278 and or Comp23 suppressed the loss of dopaminergic neurons and motor dysfunction in an animal model of 6-OHDA or rotenone-induced PD [
8,
126]. Glyoxalase activity of PARK7/DJ-1 in neuroprotection may also be of great importance since AGEs have been suggested to contribute to the development of neurodegenerative diseases. Indeed, glycation-mediated AGE formation has been reported in the Lewy bodies in PD patients [
137]. The relationship between AGEs and PD could be due to the ability of AGEs to cross-link α-syn, as has been shown using in vitro studies [
138].
4.3. Role of PARK7/DJ-1 in Alzheimer’s Disease
The role of PARK7/DJ-1 has also been suggested in AD. It was shown that the PARK7/DJ-1 binding compound UCP0054278 improved the AD-related cognitive deficits and prevented the degeneration of synaptic functions in AD modeling APdE9 transgenic mice [
9]. Similarly, a recent study by Cheng et al. demonstrated that overexpression of
DJ in the brain by lentiviral infection ameliorated β-amyloid protein (Aβ) deposition and the cognitive function of 5XFAD transgenic mice modeling AD [
139]. The results also demonstrated that reactive oxygen species and oxidative stress marker malondialdehyde content were significantly decreased, while the antioxidant superoxide dismutase activity was significantly increased in the brain of 5XFAD mice overexpressing PARK7/DJ-1 [
139]. In addition, AGEs are present in amyloid plaques in the brain of AD patients and have been suggested to promote the aggregation of Aβ and tau [
140]. According to experimental data, the glyoxalase activity of PARK7/DJ-1 may reduce these deleterious effects of AGEs in neurons. Indeed, the protective role of PARK7/DJ-1 against dicarbonyl stress was demonstrated using mouse embryonic fibroblasts, human SH-SY5Y cells, and
C. elegans, as well [
114].
4.4. Role of PARK7/DJ-1 in Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant inherited disease associated with polyglutamine expansion in the huntingtin (Htt) protein, leading to its misfolding and toxic aggregation [
141]. A recent study by Sajjad et al. demonstrated that the level of oxidized PARK7/DJ-1 Cys106 level was elevated in the frontal cortex of HD patients [
127]. They demonstrated that overexpression of PARK7/DJ-1 ameliorated mutant Htt toxicity in a yeast and Drosophila model of HD, suggesting the importance of the chaperoning activity of PARK7/DJ-1 in vivo. Their results also demonstrated that mild oxidation of PARK7/DJ-1 at cysteine 106 is required for its chaperone function; however, the complete oxidation of cysteine 106 leads to impaired PARK7/DJ-1 and detrimental cellular outcomes [
127].
4.5. Role of PARK7/DJ-1 in Ischemia-Reperfusion Induced Brain Injury
The importance of PARK7/DJ-1 has also been demonstrated in the ischemic-reperfusion injury of the brain. Indeed, intrastriatal injection of recombinant human PARK7/DJ-1 markedly reduced infarct size after middle cerebral artery occlusion of rats and protected SH-SY5Y against H
2O
2-induced apoptosis [
133]. PARK7/DJ-1-deficient animals produced a significantly larger infarct size in the animal model of Endothelin-1 induced stroke compared to wild-type controls [
142]. On the contrary, the administration of ND13 corresponding to the 13-N-terminal amino acids of PARK7/DJ-1 was shown to improve motor function after ischemic injury [
143]. Similarly, PARK7/DJ-1 binding Comp23 reduced the infarct size of cerebral ischemia in rats [
8,
136,
144].
5. Role of PARK7/DJ-1 in the Pathogenesis of Gastrointestinal Diseases
In addition to the previously discussed effects of PARK7/DJ-1 in the pathomechanism of CNS diseases, its role in the disease of other organs including the heart [
145,
146,
147], lung [
148,
149] and intestine [
10,
12,
13,
14,
114,
150,
151,
152] was recently studied.
5.1. Genetic Evidence of the Role of PARK7/DJ-1 in Gastrointestinal Diseases
First, the genome-wide association (GWA) study of Dubois et al. involving 4533 coeliac disease cases and 10750 controls suggested that the genomic region of the short arm of chromosome 1 containing the PARK7/DJ-1 gene and also that of TNF Receptor Superfamily Member 9 (TNFRSF9) is strongly associated with the risk of coeliac disease [
153]. A year later, in 2011, another GWA study comprising 6687 cases with ulcerative colitis (UC) and 19718 controls prepared by Anderson et al. revealed that the 1p36 chromosomal region, containing TNFRSF9, ERFF11, UTS2, and PARK7/DJ-1 genes, is associated with a higher risk of UC [
150].
Not long after this, Lee et al. demonstrated that cDJR-1.1, the
C. elegans homolog of the human PARK7/DJ-1 is expressed in the intestine of the worms. In addition, their results showed that lack of cDJR-1.1 makes the worms vulnerable to glyoxal-induced intestinal toxicity, giving the first in vivo evidence suggesting the protective role of PARK7/DJ-1 in intestinal pathology [
114].
5.2. Role of PARK7/DJ-1 in Coeliac Disease
The first direct human evidence suggesting the possible role of PARK7/DJ-1 in the pathomechanism of small intestinal diseases was the study of Vörös et al. [
10]. In this study, the researchers' research group demonstrated the increased mRNA expression and protein level of PARK7/DJ-1 in the small intestinal mucosa of patients with untreated coeliac disease. In this study, the researchers found that PARK7/DJ-1 immunopositivity is present in the epithelial cells of the duodenal crypt, and also in the lamina propria of duodenal biopsies derived from therapy-naive children with celiac disease. Moreover, the researchers found that, following the introduction of a gluten-free diet, the amount of PARK7/DJ-1 normalizes, suggesting the possible role of PARK7/DJ-1 in the pathomechanism of celiac disease.
5.3. Role of PARK7/DJ-1 in Inflammatory Bowel Disease
Despite the growing interest, there are relatively little data about the role of PARK7/DJ-1 in the pathomechanism of IBD. Recently, Di Narzo et al. investigated the plasma proteome of adult patients with Crohn’s disease (CD;
n = 126) and ulcerative colitis (UC;
n = 46) compared to that of healthy subjects (
n = 72) using a high-throughput SOMAmer-based capture array [
152]. They found that a total of 493 proteins showed altered levels in the plasma of patients with IBD compared to healthy subjects; among them, 219 were up- and 274 were down-regulated. One identified protein with an increased presence in the plasma of UC patients compared to that of healthy subjects was PARK7/DJ-1.
The role of PARK7/DJ-1 in coeliac disease and IBD-related intestinal inflammation is demonstrated, however, further studies are needed to resolve the contradictions and elucidate the precise role of PARK7/DJ-1 in IBD.
5.4. The Role of PARK7/DJ-1 in Intestinal Dysbiosis
A recent publication of Singh et al. investigated the effect of PARK7/DJ-1 deficiency on the intestinal microbiome of healthy mice [
14]. They investigated the effect of PARK7/DJ-1 deletion on bacterial composition of the intestine by 16S rRNA sequencing. Analysis of fecal samples showed that overall composition of the microbiome did not differ between PARK7/DJ-1−/− and PARK7/DJ-1+/+ and mice at the phylum level. However, calculation of the F/B ratio showed that it decreased significantly in PARK7/DJ-1−/− mice compared to PARK7/DJ-1+/+ mice, suggesting the functional role of PARK7/DJ-1 on the composition of the intestinal microbiome. The deeper analysis of the data showed an increased presence of Alistipes and Rikenella species in PARK7/DJ-1−/− mice. The role of these species has been described regarding the pathomechanism of IBD [
159].
Changes in the composition of intestinal microbiome affect its metabolite production. Accordingly, they demonstrated that the amount of fecal and also that of serum amino acids, including valine, leucine, phenylalanine, alanine, tyrosine and isoleucine, were downregulated, whereas SCFAs, including malonate, dimethylamine, trimethylamine and acetoin, were upregulated in PARK7/DJ-1−/− mice compared to that of PARK7/DJ-1+/+ mice. Although the metabolic changes of PARK7/DJ-1−/− mice were complex, Singh et al. suggested that it can lead to metabolic stress of the intestine, as demonstrated by the increased inflammation of the intestine. Indeed, they found increased levels of pro-inflammatory monocyte chemotactic protein-1 (MCP-1) and calprotectin in feces of PARK7/DJ-1−/− mice compared to that of PARK7/DJ-1+/+ mice. However, it must be also noted that the expression of other pro-inflammatories such as granulocyte monocyte colony stimulating factor IFN-β decreased, and that of many others, including IL-12p70, TNF-α, IL-17A, IFN-γ, IL-23 and IL-6 did not change.
Finally, since the association of the intestinal microbiome and also that of PARK7/DJ-1 with neurodegenerative diseases is well known, they investigated the molecular biological changes in the midbrain of PARK7/DJ-1−/− mice by RNA-sequencing. They found the upregulation of PD related inflammatory genes, including polymerase family member 1 (Parp1) and MMP-8 in the midbrain of PARK7/DJ-1−/− mice compared to that of PARK7/DJ-1+/+ mice, suggesting that changes in the intestinal microbiome and/or lack of PARK7/DJ-1 may alter the pathology of the CNS.
6. Role of PARK7/DJ-1 in GBA Diseases
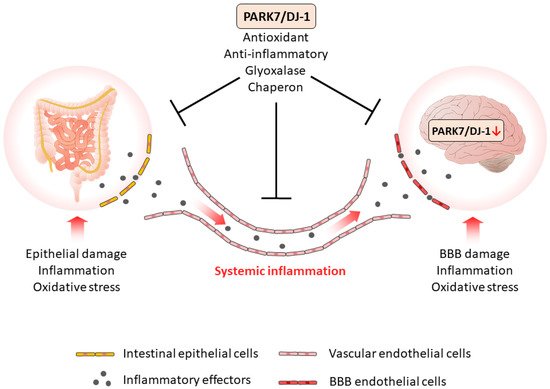
Based on the current knowledge, it is assumed that PARK7/DJ-1 represents a molecular link between intestinal and brain diseases. Indeed, recent studies demonstrated that PARK7/DJ-1, through its antioxidant and anti-inflammatory properties, plays a role in the maintenance of healthy intestinal microbiome and mucosal integrity, thus influencing the local and systemic inflammation characteristic for IBD.
As intestine-derived inflammatory factors can reach the BBB and impair its integrity, the inflammation can also spread also to the brain. The increased presence of inflammatory mediators induces the inflammation of CNS and may also alter the synthesis and function of PARK7/DJ-1 itself in the brain (Figure 1). Indeed, it has been shown that TNF-α, TGF-β, or LPS reduces the expression of PARK7/DJ-1, and also that increased amounts of MMP-3 may induce its degradation (Table 2). Also, the oxidative stress is an important factor that regulates the synthesis and function of PARK7/DJ-1 in the brain. Moreover, oxidative stress has been demonstrated to play a key role in the inactivation and degradation of PARK7/DJ-1 (Table 2).
Figure 1. The role of PARK7/DJ-1 in GBA diseases. PARK7/DJ-1, through its antioxidant, anti-inflammatory, glyoxalase, and chaperon activity properties, plays a role in the maintenance of intestinal integrity, thus diminishing the local and the systemic inflammation. The systemic inflammation of intestinal origin can be considered as a possible factor that negatively regulates PARK7/DJ-1 in the brain. Similarly to the gut, PARK7/DJ-1 also plays a protective role in the brain, therefore, its negative regulation enhances pathological GBA crosstalk and the development of neurodegenerative diseases.
Table 2. Molecular regulation of PARK7/DJ-1.
|
Molecule
|
Effect on PARK7/DJ-1
|
Tissue or Cell Type
|
Refs.
|
|
Negative regulators of PARK7/DJ1
|
|
H2O2
|
Overoxidation
|
Human brain
|
[80,81]
|
|
p53
|
Reduced expression
|
mouse embryonic fibroblasts
|
[91]
|
|
BAG5
|
Decreased stability
|
HEK293 human embryonic kidney
|
[92]
|
|
MMP-3
|
Proteomic fragmentation
|
CATH.a mouse neuronal
|
[93]
|
|
LPS
|
Reduced expression
|
HT-29 human colonic adenocarcinoma
|
[11]
|
|
TNF-α
|
Reduced expression
|
HT-29 human colonic adenocarcinoma
|
[11]
|
|
TGF-β
|
Reduced expression
|
HT-29 human colonic adenocarcinoma
|
[11]
|
|
miR-128-3p
|
Reduced expression
|
Human hepatocellular carcinoma
|
[94]
|
|
miR-494
|
Reduced expression
|
3T3-L1 mouse adipocytes and Neuro-2a neuroblastoma
|
[96]
|
|
miR-203
|
Reduced expression
|
SW1990/DDP human pancreatic cancer cells
|
[95]
|
|
Positive regulators of PARK7/DJ1
|
|
STAT5A
|
Increased expression
|
human leukemic pre-B
|
[97]
|
|
SG2NA
|
Protection from degradation
|
Neuro2a neuroblastoma
|
[99,100]
|
|
IL-17
|
Increased expression
|
HT-29 human colonic adenocarcinoma
|
[11]
|
|
H2O2
|
Increased expression
|
HT-29 human colonic adenocarcinoma
|
[11]
|
PARK7/DJ-1 similarly to its effects in the gut has great importance in the protective mechanisms of the brain. Therefore, molecular mechanisms that alter PARK7/DJ-1 activity may contribute to the development of neurodegenerative disorders (Figure 1).
Although the function and regulation of PARK7/DJ-1 are still not completely understood, there is a growing body of evidence suggesting that it participates in the pathomechanism of diseases influenced by GBA.