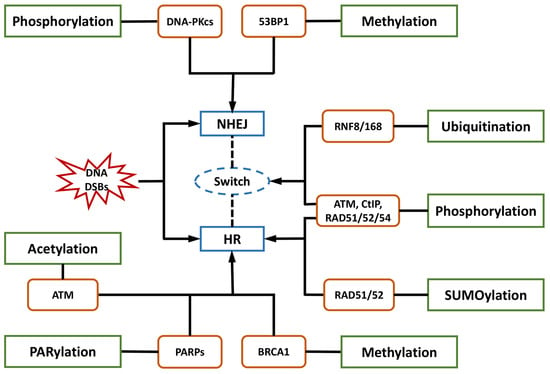
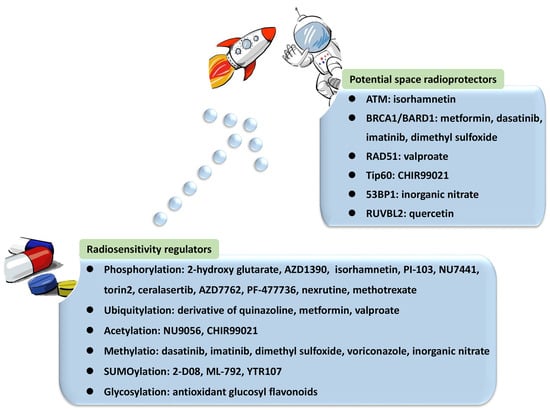
Table 1. Essential factors in post-translational modifications (PTMs) in repair of DNA double-strand breaks (DSBs), their cellular functions, and application in radiosensitization and radioprotection.
3. Targeting Essential Phosphorylation Factors for Regulating DDR
The essential participants in phosphorylation, which regulates the alterations of protein conformation and biological activity, include ATM, DNA-PKcs, CtIP, ATR, CHK1, H2AX, and RAD51/52/54.
ATM is one of the major and most extensively studied kinases involved in the early stages of cellular responses to DNA DSBs [
86]. ATM is activated in the phosphorylation of H2AX [
86,
87]. Many ATM-dependent phosphorylation processes need sustained activity of ATM, and a phosphorylation site on ATM itself functions in its retention on damaged chromatin [
159]. ATM mediates the rapid phosphorylation of DNA-PKcs at Thr-2609 and the adjacent (S/T) Q motifs within the Thr-2609 cluster upon IR-induced DNA DSBs [
88], as well as MDM2 phosphorylation at multiple sites near its RING domain [
89]. In contrast, mutation at lysine 3016 of ATM inhibits the phosphorylation of p53 and CHK2 [
90], which was reported to play a key role in stabilizing p53 [
89]. ATM has been considered a drug target because increased ATM is related to the development of radioresistance [
160]. Thus, this kinase has been frequently investigated in attempts to improve radiation therapy (RT) in cancer treatment [
161,
162]. It is inhibited by radiosensitizers [
163] such as 2-hydroxy glutarate [
91] and AZD1390 [
92]. It is also inhibited by caffeine, according to DrugBank [
164]. A considerable number of ATM inhibitors are under development as components of future cancer therapies [
165]. A radioprotector named isorhamnetin, targeting ATM, was reported in 2021. It prevents IR-induced gastrointestinal syndrome by promoting ATM phosphorylation and enhancing the recruitment of 53BP1 [
93].
An important partner of ATM is DNA-PKcs. DNA-PKcs phosphorylation mediated by ATM is needed for the full activation of DNA-PKcs and subsequent DSB repair [
88]. DNA-PKcs targets phosphorylation sites on ATM, and phosphorylation mutations markedly inhibit ATM activity and affect the ATM signaling of DSBs [
166]. DNA-PKcs is highly relevant to cancer. Previous studies reported its involvement in cancer metabolism by impacting glycolysis [
167], emphasizing it as a promising therapeutic target. Interestingly, different regulatory co-factors induce distinct DNA-PKcs phosphorylation kinetics at Thr-2609 and Ser-2056, playing primary roles in DSB repair and the establishment of cellular radioresistance [
88]. Thus, DNA-PKcs is often targeted for overcoming radioresistance in cancer therapies. For example, the triple-target (DNA-PK/PI3K/mTOR) inhibitor PI-103 was found to increase the radiosensitivity of a glioblastoma cell line subtype by targeting DNA-PKcs [
94]. The sensitivity of several other kinds of cancer cells to RT and chemotherapy could be significantly promoted by NU7441 as a DNA-PKcs inhibitor [
95,
96]. According to DrugBank, DNA-PKcs is targeted by caffeine and SF1126. The latter is an investigational agent at present for the treatment of various forms of cancers. Decreases or deficiencies in DNA-PKcs lead to accelerated cellular senescence [
168], which offers clues for protecting cells from radiation-induced premature senility.
One of the sophisticated molecular switches controlling the balance between NHEJ and HR in DSB repair is the phosphorylation of serine 327 in CtIP throughout the cell cycle [
42]. DNA end resection was found to occur not only during HR but also in NHEJ in the G1 phase in a distinct manner controlled by CtIP [
97]. In this process, CtIP is phosphorylated by PIK3 and interacts with BRCA1, stimulating the initiation of resection [
169]. CtIP is among the proteins participating in the resections of the late and persistent DSBs. CtIP is made use of in cancer treatment, since it increases p38-MAPK reactivation in cooperation with CHK1 in response to RT [
170]. The ATP-competitive mTOR inhibitor torin2 reduces the number of radiation-induced CtIP and RAD51 foci formed, indicating that this compound radiosensitizes cancerous tumors by blocking HR. Mechanically, it causes an S-phase-specific DNA repair deficiency [
99]. Regarding radioprotection, CtIP was reported to function in DSB repair by NHEJ in the G0/G1 phase through phosphorylation at Thr-847 [
171]. The engagement in DNA repair implied the potential use of CtIP in preventing and relieving radiation injuries.
ATR and CHK1 are common participants in phosphorylation. ATR phosphorylates several BRCA1 fragments directly in response to IR [
100]. Interactions between these two proteins have also been observed. The ATR-CHK1 pathway was implicated in DDR processes [
172]. CHK1 phosphorylation at serine 345 is suppressed by the overexpression of ATR in mutated cells and enhanced in wild-type cells [
173]. ATM and the nuclease activity of MRE11 are required for ATR recruitment, followed by the phosphorylation and activation of CHK1 [
174]. These two proteins have been identified as ideal therapeutic targets in cancer treatment, and their inhibitors have been in clinical trials either as single drugs or in combination with other genotoxic agents [
175]. A growing number of research studies proved the efficiency of ATR inhibitors as viable anticancer drugs. Eight of them are currently under development [
101], among which ceralasertib is included in DrugBank. These kinds of inhibitors were shown to be effective, especially in the treatment of head and neck squamous cell carcinoma, in combination with other therapies such as surgery, RT, and chemotherapy [
176]. They also specifically target DDR pathways and impair DSB repair processes, exhibiting significant radiosensitizing effects [
177]. CHK1 is also related to as many as thirty-eight compounds, according to DrugBank, the majority of which are investigational or experimental. The only approved drug is fostamatinib, which is used in the treatment of chronic immune thrombocytopenia. Nevertheless, targeting CHK1 represents an attractive strategy for potentiating the efficacy of RT. DDR proteins, including CHK1 and ATR that promote HR, are upregulated in radioresistant breast cancer cells, and the CHK1 inhibitor AZD7762 sensitizes these cells to IR [
98]. Another CHK1 inhibitor, PF-477736, was proven to enhance the radiosensitivity of human triple-negative breast cancer cells [
102]. Nexrutine was reported to increase the sensitivity of prostate cancer cells to IR by reducing the expression of several proteins, including CHK1 [
103]. However, no radioprotectors have yet been developed based on ATR or CHK1.
RAD51 and its paralogs function in the transduction of DNA damage signaling and facilitate the repair of DNA breaks [
43]. RAD51 is believed to be among the central factors in HR repair and regulated at the level of PTM in this pathway. The most significant PTM related to RAD51 is phosphorylation, which mediates distinct functions in promoting HR in response to global DNA damage [
37]. Phosphoaminophosphonic acid-adenylate este and amuvatinib were collected in DrugBank targeting RAD51. The latter is in trials for treatment of solid tumors. Evidence shows that targeting RAD51 contributes to overcome the radioresistance of some kinds of cancers [
70,
104,
105], such as methotrexate inhibiting RAD51 expression and radiosensitizing human osteosarcoma cells [
106]. A RAD51 paralog, RAD52, was implicated in phosphorylation at T412, facilitating later stages of HR [
37,
178]. Its enhancement elicits the radioresistance of cancer stem cells [
107] and it was widely explored in synthetic-lethality-based anticancer therapies [
179]. RAD54, which participates in polymerase-dependent DNA synthesis and the completion of HR [
180,
181], was found to be phosphorylated by NEK1 during the G2 phase and by CDK2 to limit its branch migration activity in HR [
37,
182]. However, reports on its use in radiosensitivity regulation are insufficient.
Histone H2AX is one of the regulators of the checkpoint pathways responding to DNA DSBs [
108]. It is mainly phosphorylated by ATM at serine 139 in the early stages of DNA DSBs and forms foci at the sites of DNA damage [
86,
183]. H2AX phosphorylation contributes to DDR, and the ability to enhance H2AX phosphorylation decreases substantially in DNA-repair-deficient cells after IR [
183]. H2AX also facilitates 53BP1 recruitment to DNA break sites [
109]. However, there are few reports on the application of H2AX in regulating radiation-induced DDR.
Other crucial factors in phosphorylation include BRCA1, CHK2, and the MRE11-RAD50-NBS1 complex (MRN), as well as the downstream effectors p53, NF-κB, AKT, and survivin [
184]. In addition, bioprocesses such as autophosphorylation [
47,
185], hyperphosphorylation [
100,
186], and dephosphorylation [
159] all play important roles in the regulation of the multi-protein network, which is irreplaceable for the maintenance of genomic integrity [
187]. An extensive overview and deep understanding of them may contribute to an elucidation of the detailed molecular mechanisms of cellular DDR to different kinds of radiation and the discovery of novel regulators by targeting signal pathways related to phosphorylation.