Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Bacteria can exhibit two types of antibiotic resistance: intrinsic and acquired. While intrinsic resistance is determined by naturally occurring mechanisms conferred by inherent structural and/or functional features of the bacteria, the acquired resistance results from the changes in the bacterial genome. These consist of mutations in antibiotic-targeted genes or the acquisition of exogenous DNA conferring resistance, horizontally transferred by plasmids, bacteriophages, transposons, or other mobile genetic elements. Many independent mechanisms of bacterial resistance to antibiotics have been identified, including primarily modification of the antibiotic target, changes in the cell envelope’s permeability, active pumping of the antibiotic out of the cell (so-called efflux system), and enzymatic inactivation.
- antibiotic resistance
- antibiotics
- bacteria
- mechanism of action
1. Modification of the Antibiotic Target Site
The resistance determined by a modification of the target site of an antibacterial substance action constitutes a large and heterogenous group of mechanisms of a different mode of action.
In the case of β-lactam-resistant bacteria, the resistance is based on a modification of the structure of natural PBP proteins and has been best described in methicillin-insensitive Staphylococcus aureus (MRSA) expressing modified PBP2a (also called PBP2′) [1][2]. PBP2a transpeptidases have a reduced affinity for β-lactams but, at the same time, retain catalytic functions [3]. The altered PBPs group also includes PBP1a, PBP2b, and PBP2x enzyme types observed most frequently in Staphylococcus pneumoniae, or PBP5 and PBP3r observed in Enterococcus hirae S185 isolates [4][5]. The mecA and mecC genes, which encode PBP2a transpeptidases, determine resistance to almost all β-lactam antibiotics, except cefaroline and ceftobripol. They are located together with the regulatory genes mecI and mecR1 within the staphylococcal SCCmec chromosomal cassette integrated into the bacterial chromosome [6][7]. In contrast, the mecB gene that determines similar β-lactam antibiotic resistance in Staphylococcus aureus was identified in the large plasmid pSAWWU4229_1 [8]. Furthermore, a new mecD gene has been detected in Macrococcus caseolyticus isolates within so-called genomic resistance island. It has been shown that mecD confers resistance to all β-lactam antibiotics, including cephalosporins, ceftaroline, and ceftobiprole [9] (Figure 1(1)).
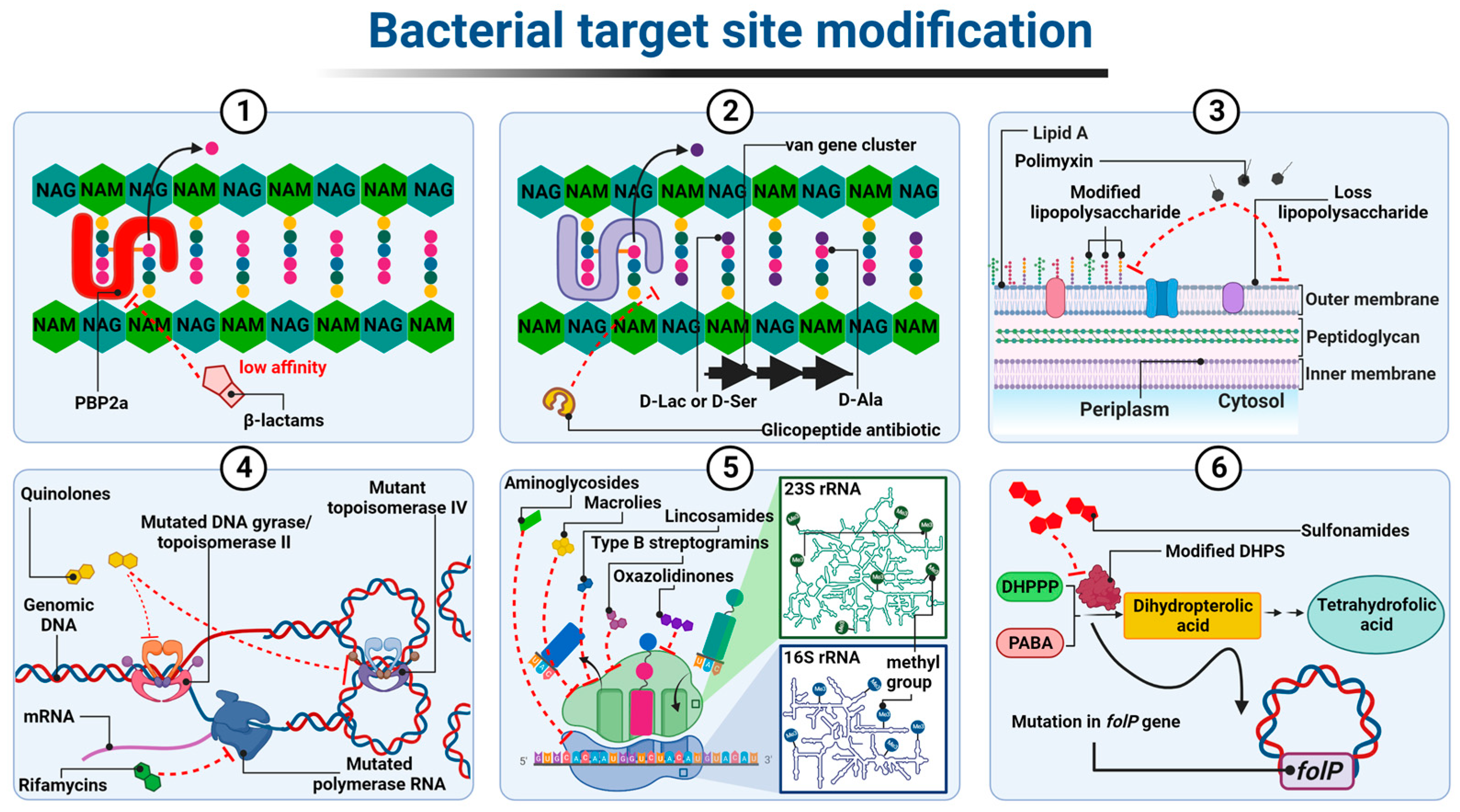
Figure 1. Diagrammatic illustration of some possible resistance mechanisms based on target site modification of antibiotics: (1) alteration in PBP; (2) altered cell wall precursors; (3) modified or loss of lipopolysaccharide; (4) mutated DNA gyrase/topoisomerase IV or RNA polymerase; (5) alteration in the 30S or 50S subunit; (6) modified DHPS. Figure created with Biorender.com.
The resistance to glycopeptides is determined by the synthesis of altered peptidoglycan precursors, which end in carboxyl-terminated D-alanyl-D-lactate (D-Ala-D-Lac) or D-alanyl-D-serine (D-Ala-D-Ser) The incorporation of the D-Lac residue into the Lipid II chain prevents the formation of one out of the five hydrogen bonds that connect Lipid II to the glycopeptide molecule, resulting in an approximately 1000-fold decrease in the affinity of the antibiotic for D-Ala-D-Lac. In contrast, the substitution of D-Ser in place of one D-Ala at the precursor end of the pentapeptide leads to an approximately seven-fold decrease in the binding strength of the glycopeptide to D-Ala-D-Ser, probably due to spherical effects [10][11][12]. The ability to express drug-resistant murein precursors is determined by the presence of Van operons, (i.e., VanA, VanB, VanC, VanD, VanE, VanD, VanG, VanL, VanM, and VanN) in the bacterial genome [13]. It should be noted that the VanA and VanB operons (conferring resistance to mainly vancomycin and teicoplanin or only to vancomycin, respectively) have the highest prevalence in the bacterial world. As they are encoded by genes located in transposons, they can be transferred between different bacterial species using plasmids or chromosome fragments [14][15][16] (Figure 1(2)).
Resistance to antibiotics that alter the integrity of the outer membrane, i.e., polymyxins, is generally the result of structural modifications of LPS that involve the attachment of positively charged molecules of 4-amino-4-deoxy-L-arabinose (L-Ara4N), phosphoethanolamine (PEtN), or galactosamine to the phosphate groups of lipid A [17]. As a result, the negative charge of lipid A is reduced, which leads to the inhibition of the electrostatic interactions of polymyxin molecules with this component. The synthesis of LPS containing L-Ara4N groups is an innate characteristic of Burkholderia spp., Proteus spp., and Chromobacterium violaceum [18][19][20]. In other Gram-negative bacteria, structural modification is mediated by two major two-component regulatory systems, PhoP-PhoQ and PmrA-PmrB. The individual system comprises a sensor histidine kinase, PhoQ or PmrB, and a response regulator, PhoP and PmrA, respectively. The role of the latter is to control gene expression. Both systems have been identified in Salmonella enterica, Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, and Yersinia spp. [21][22][23][24][25]. Under conditions such as low concentrations of Mg2+ and Ca2+, low pH, or in the presence of polymyxins in the environment, sensor histidine kinase, that is PhoQ or PmrB, trans-autophosphorylates within its dimer. The phosphate is then transferred to response regulators, that is PhoP or PmrA, respectively. The latter positively regulate the transcription of genes responsible for the modification of lipid A in LPS [26].
Furthermore, mutations in genes that encode proteins in these regulatory systems lead to their constitutive activation, which is reflected in the overexpression of genes independently of environmental stimuli and an increased resistance level to polymyxins [27].
Of note, Acinetobacter baumannii has been confirmed to be the only known LPS-deficient strain to date, due to mutations within the lpxA, lpxC, and lpxD genes, whose products catalyze the initial steps of lipid A biosynthesis [28]. Thus, the polymyxin molecule cannot anchor within the Acinetobacter baumannii outer membrane, resulting in cell insensitivity to antibiotics (Figure 1(3)).
The resistance to quinolone antibiotics involves remodeling the chemical structure of gyrase and/or topoisomerase IV, which becomes insensitive to the drug. These structural changes are driven by point mutations in chromosomal genes encoding the gyrase and topoisomerase IV subunits, particularly the GyrA and ParC ones, respectively. They occur within specific codons located in the so-called quinolone resistance determinant region, QRDR within gyrA, gyrB (encoding gyrase subunits), parC, and parE (encoding topoisomerase IV subunits) sequences. In the amino acid sequence of the protein, these regions correspond to stretches comprising domains at the amino end of both subunits that are adjacent to the tyrosine residues of their active site [29][30].
There is one exception, however, as serine substitutions have been shown not to affect the efficiency of both topoisomerases II, even in the presence of antibiotics. In addition to the most frequent mutations within gyrA and parC sequences, mutations that also occur in genes that encode GyrB and ParE subunits of gyrase and topoisomerase IV, respectively, can confer resistance to quinolones [31][32]. Notably, in some species—for example, Staphylococcus aureus, Streptococcus pneumoniae, or Salmonella spp.—a high level of resistance to quinolones is conferred by several mutations that occur simultaneously in genes encoding both gyrase and topoisomerase IV [33][34][35].
The resistance to rifampicins is related to conformational changes in the β-subunit of DNA-dependent RNA polymerase (RNAP) (resulting in the loss of its affinity for antibiotic) determined by mutations in the rpoβ gene (rarely rpoC and rpoA encoding α subunit of RNAP) [36][37][38][39][40][41][42]. (Figure 1(4)). Among the mechanisms underlying bacterial resistance to antibiotics, which act on the 50S ribosome subunit, are those executed by post-transcriptional modifications of 23S rRNA nucleotides resulting from direct methyltransferase activity. In the case of macrolides (but only those with a 14- or 15-member lactone ring), lincosamides, and streptogramins B, the aforementioned strategy is associated with the presence of methyltransferases encoded by erm genes. These enzymes, using S-adenosyl-L-methionine as a donor methyl group, catalyze the monomethylation or dimethylation reaction of the N6 atom of adenine A2058 (Escherichia coli numbering) within the PTC 23S rRNA. Subsequently, this inhibits antibiotic binding to this nucleotide [43][44]. Among the Erm methyltransferases described to date, the ErmC class is the most prevalent in staphylococci (e.g., Staphylococcus aureus), while the ErmB and ErmA classes are the most common in enterococci (e.g., Enterococcus faecalis) and streptococci (e.g., Streptococcus pneumoniae) [45][46][47][48]. Erm genes in most microorganisms are located within the transposons and, as a mobile genetic element, they are involved in the spread of the so-called MLSB-type resistance [49][50][51]. In linezolid-resistant bacteria, plasmid-encoded methyltransferases, products of the cfr gene, are responsible for the modification of 23S rRNA by adding a methyl group to the C8 atom of its A2503 adenine (Escherichia coli numbering). This mechanism, together with the loss of sensitivity to oxazolidinones, leads bacteria to acquire resistance to lincosamides, streptogramins A, macrolides with a 16-member lactone ring, pleuromutilins, and phenicols [52][53].
Moreover, the rRNA within the 30S subunit of the ribosome can be methylated in order to protect bacterial cells from bactericidal effect of aminoglycosides. For example, ArmA, a methyltransferase that was primarily identified in Enterobacteriaceae and Acinetobacter baumannii species, methylates the N7 guanine atom of the G1405 16S rRNA and, consequently, determines high levels of resistance to gentamicin, tobramycin, and amikacin [54].
In addition to rRNA methylation, mutations in genes encoding ribosomal proteins and changes in 16S and 25S rRNA sequences also lead to a decrease in the affinity of protein biosynthesis inhibitors for both ribosomal subunits [55][56]. However, mutations that occur in genes encoding 16S rRNA do not play an important role in bacterial resistance to aminoglycosides, because such molecular changes (with the exception of the A1408G substitution) generally cause bacterial cell death [57][58] (Figure 1(5)).
Modification of the target of an antimicrobial substance is one of the most important mechanisms of the bacterial acquisition of resistance to sulfonamides. Numerous studies have shown that drug-resistant strains produce modified DHPS due to mutations within the conserved regions of the chromosomal folP gene (sulA). These mutations result in reduced drug sensitivity DHPS. At the same time, drug affinity for PABA is maintained or even increased. In Neisseria meningitidis and Streptococcus pneumoniae mutants, this type of resistance results from the insertion of six-base-pair-long sequences encoding two additional amino acids in drug-resistant synthetase (Figure 1(6)).
2. Changes in the Permeability of a Bacterial Cell
The mechanism leading to changes in bacterial cell permeability is commonly employed by Gram-negative bacteria because the structure of their cell wall (when compared to that of Gram-positive bacteria) allows greater regulation of the substance penetration into the cell [59][60][61][62][63][64]. Reduced outer membrane permeability increases bacterial resistance to drugs and is particularly conferred by changes in the qualitative composition of porins, alterations in their functionality or selectivity, as well as in a decrease in porin-encoding gene expression [65].
For example, Klebsiella pneumoniae possess two main general diffusion porins, Ompk35 and Ompk36, through which antibiotics, i.e., β-lactams and fluoroquinolones, can enter the cell [66]. The reduced sensitivity of most strains to cefotoxam and cefoxitin, but not carbapenems, is caused by the loss of Ompk35 and Ompk36 porins, which is associated with the simultaneous synthesis of Ompk37 porins with a narrow diffusion channel [67]. On the contrary, the lack of expression of genes encoding both of these porins—which is, generally, driven by point mutations or insertional rearrangements throughout in their coding or promoter sequences—results in the acquisition of resistance to cephalosporins and carbapenems [66][67][68].
Another example is the carbapenem-resistant Pseudomonas aeruginosa strains, whose resistance to carbapenems (i.e., imipenem and meropenem) is associated with negative transcriptional regulation or mutations in the oprD gene that directly inhibits OprD porin synthesis [62][69][70]. Another mechanism is based on a modification of the function of the WalKR two-component regulatory system. This system is characteristic of Gram-positive bacteria with a low content of GC pairs in their genomes. It consists of the histidine kinase WalK and the response regulator WalR, whose orchestrated action alters the expression levels of genes under their control. One such example is genes involved in the regulation of cell wall metabolism [71], such as glmU and murG [72][73]. Their overexpression, which is driven by mutations in genes that encode components of the regulatory system and/or its upstream elements, results in synthesizing a cell wall with a higher peptidoglycan content [72][73]. The increased thickness of the cell wall makes it more difficult for antibiotic molecules to enter the cell, which contributes to the reduced sensitivity of staphylococci to antibiotics (Figure 2).
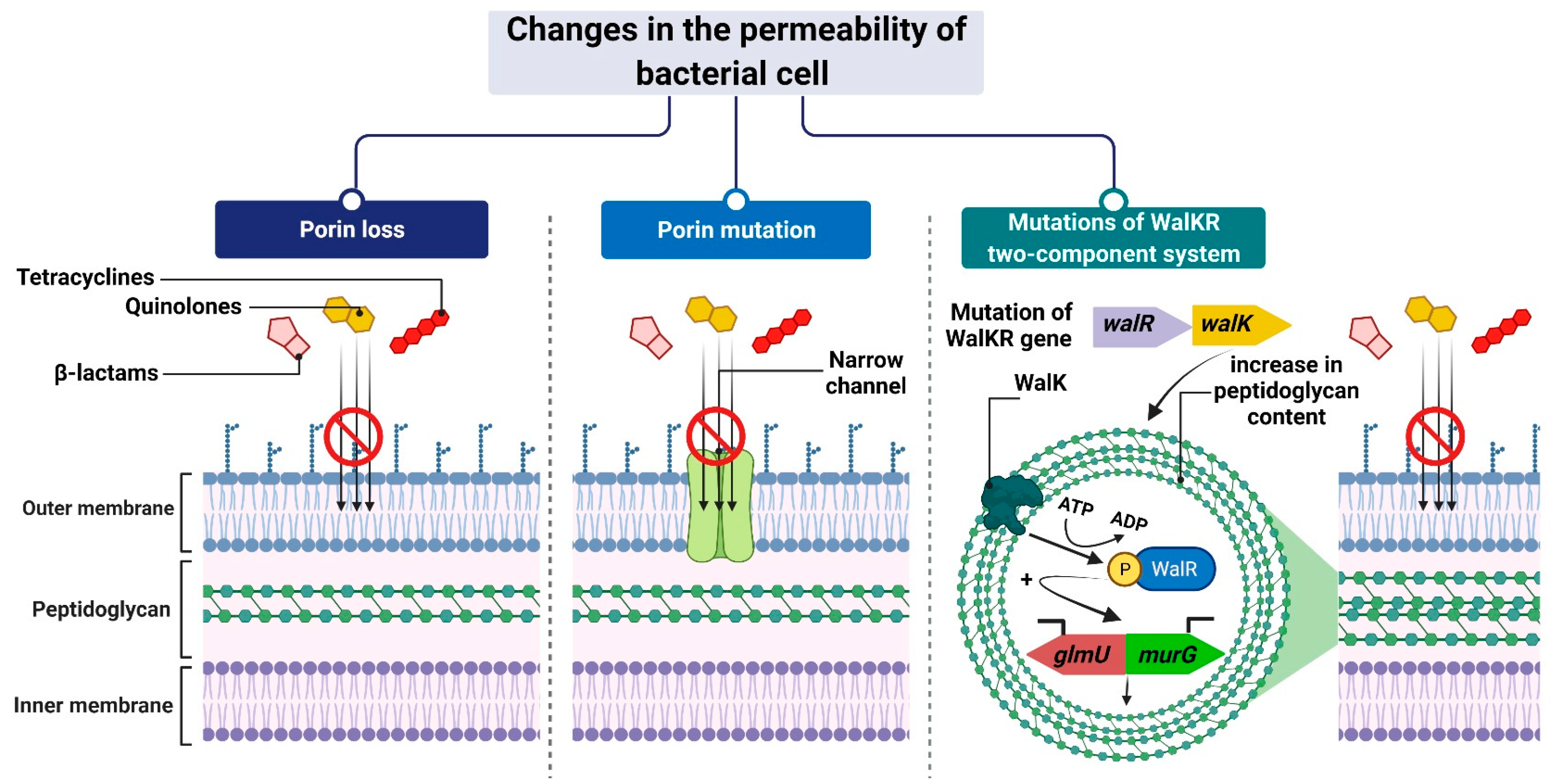
Figure 2. Reduced antibiotic accumulation through changes in the permeability of the bacterial cell. Figure created with Biorender.com.
3. Active Pumping of the Antibiotic out of the Cell
One common mechanism of drug resistance is the active efflux of drugs from bac-terial cells to prevent the intracellular accumulation of toxic compounds. Drug-resistant bacteria contain energy-driven drug efflux pumps that squeeze out antibacterial agents, thereby reducing their intracellular concentrations in a way that does not involve alteration or degradation [74].
These pumps, encoded by genes located on chromosomes, in mobile genetic parts (MGEs) or plasmids, differ in, e.g., their structures, substrate spectrum, and source of energy necessary for transport. Therefore, they are divided into six families: MFS (major facilitator superfamily), SMR (small multidrug resistance family), PACE (proteobacterial antimicrobial compound efflux), MATE (the multidrug and toxic compound extrusion family), ABC (ATP-binding cassette superfamily), and RND (the resistance nodulation division family) [75] (Figure 3).
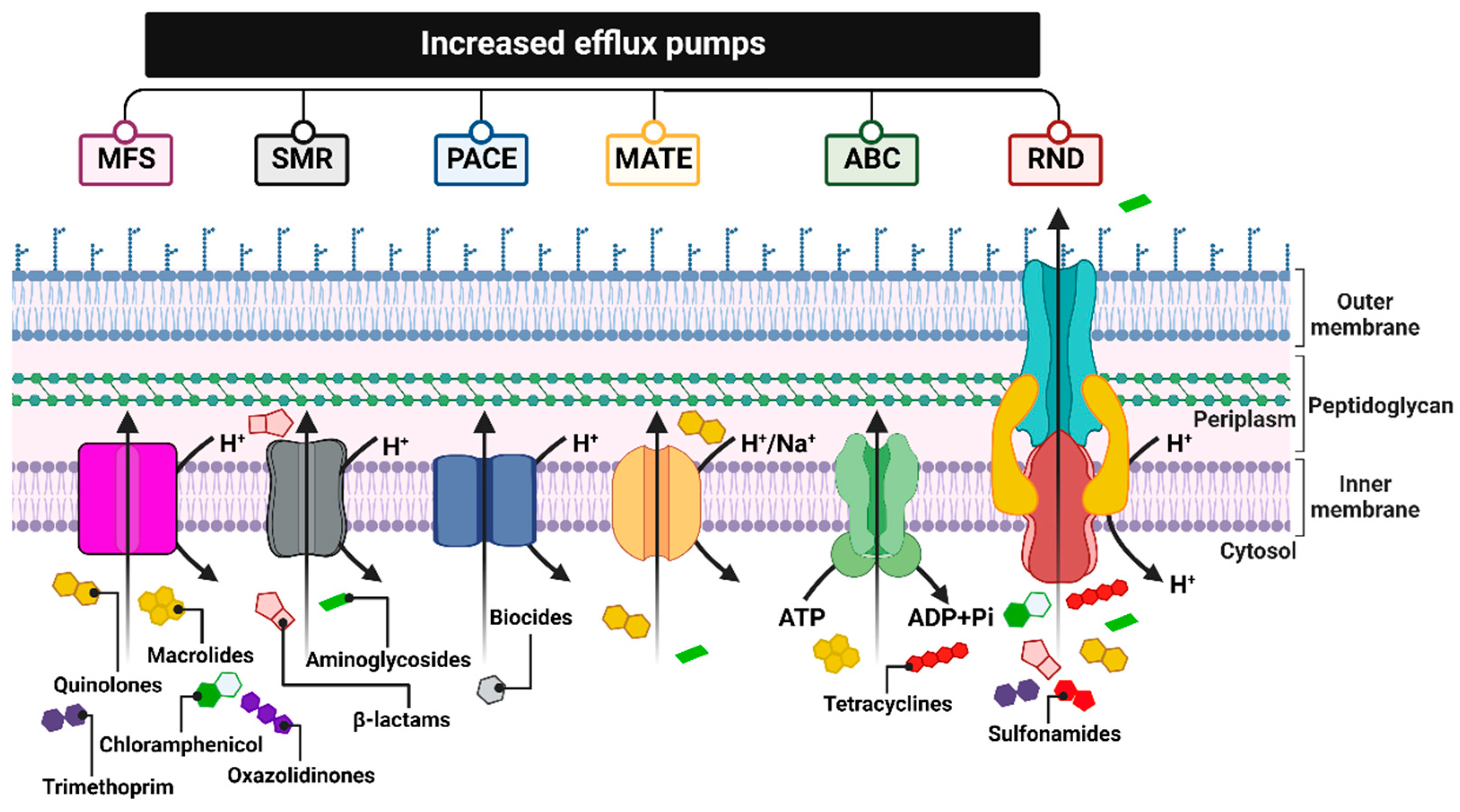
Figure 3. Summary of the six major families of efflux transporters: MFS (a superfamily of the main facilitator), SMR (the small multidrug resistance family), PACE (proteobacterial antimicrobial compound efflux), MATE (multidrug and toxic compound extrusion family), ABC (ATP binding cassette superfamily) and RND (resistance nodulation division family). Figure created with Biorender.com.
MSF family proteins are responsible for conferring resistance to fluoroquinolones, macrolides, chloramphenicol, linezolid, trimethoprim, and others. One of the examples is: NorA pumps identified in Staphylococcus aureus and MefB observed in Escherichia coli strains [76].
SMRs, a small multidrug resistance family, are involved in reducing the susceptibility of bacterial cells mainly to β-lactams and some aminoglycosides, as confirmed in Escherichia coli (EmeR pump) and Staphylococcus epidermidis (SMR pump) isolates [75][77].
The PACE pumps identified in the Acinetobacter baumannii isolates probably form four trans-membrane α-helices and are composed of 150 amino acids. Their substrate spectrum is limited to commonly used biocides, i.e., chlorohexidine, acriflavine, benzalkonium, or proflavine [75][78][79].
MATE-type pumps, acting as antiporters, derive the energy necessary for their activity from the hydrogen or sodium ion gradient. The MATE proteins contribute to the reduced efficacy of fluoroquinolone and some aminoglycoside antibiotics [80]. Among the representatives of this transporter family are: NorM pumps, which are found in Neisseria gonorrhoeae [81], and MepA, identified in Staphylococcus aureus.
Another family of multidrug efflux pumps is the ATP-binding cassette (ABC) family, which operates through energy derived from the hydrolysis of ATP molecules [82].
ABC pumps, for example, MacB found in Escherichia coli, allow bacteria to actively transport antibiotics such as tetracyclines and macrolides outside the cell [83].
Among the different types of efflux pumps, the resistance nodulation division (RND) superfamily is considered the main drug efflux pumps family, as it confers drug resistance to various species of Gram-negative bacteria.
RND pumps, as well as some ABC, MATE, and MFS pumps, form three-membered protein structures that are located within the entire bacterial cell membrane. The transport proteins of these systems are embedded in the inner membrane (cytoplasmic) and interact with the proteins acting as channels in the outer membrane, as well as acting as the fusion proteins of the periplasmic space (connecting the two membranes), removing antibiotic molecules directly to the external environment. This mechanism of action makes it difficult for the antibiotic to return to the bacterial cell. On the contrary, most MSF and SMR pumps consist of a single protein transporter located in the cytoplasmic membrane, which pumps antibiotic molecules only into the periplasmic space, thus allowing them to easily return to the cytosol. RND pumps, which serve as substrate/H+ ion antiport, are characterized by a broad spectrum of transported substrates [84]. The aforementioned pump systems can contribute to multidrug resistance, especially to tetracyclines, chloramphenicol, β-lactams, aminoglycosides, quinolones, sulfonamides, or trimethoprim [85]. Among the RND pump systems identified so far, which, nota bene, are observed only in Gram-negative bacteria, the best known are two ternary complexes: MexAB-OprM in Pseudomonas aeruginosa [86] and AcrAB-TolC, which occur in many species of the Enterobacteriaceae family, including Escherichia coli, Salmonella enterica serovar Typhimurium, or Klebsiella pneumoniae [32].
The transport of substances through the efflux system is effectively controlled by local regulatory proteins (e.g., BmrR found in Bacillus subtilis), as well as global cellular regulatory proteins (e.g., MarR in Escherichia coli) [87]. The overexpression of efflux pumps causes an above-average increase in the efficiency of antibiotic elimination from the bacterial cell and usually results from mutations (deletions, insertions) within the genes encoding these regulatory proteins.
4. Enzymatic Inactivation
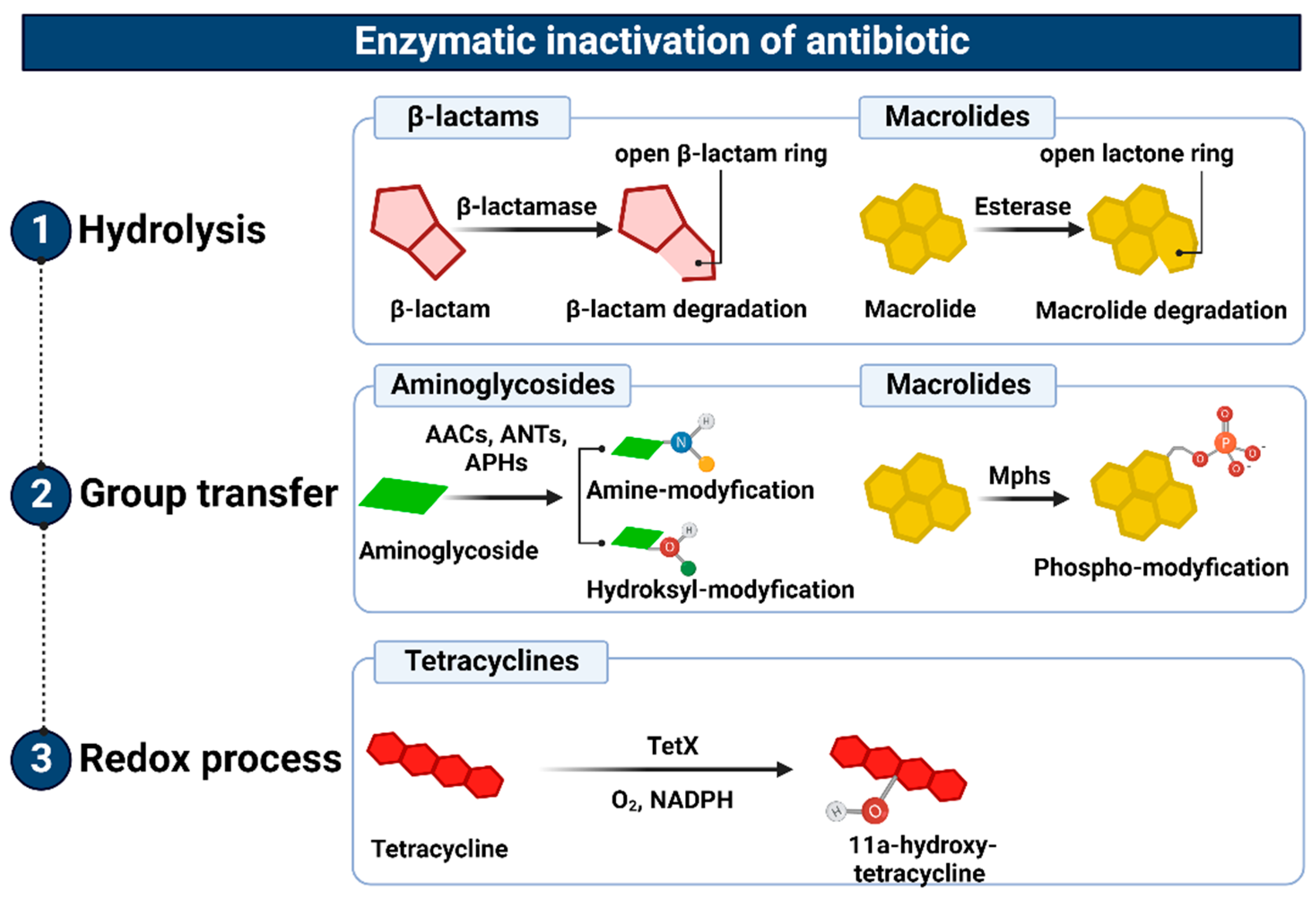
The enzymatic inactivation of antibiotics can be executed by hydrolysis, group transfer, or redox process [88]. In the case of β-lactam antibiotics, resistance is mediated by β-lactamases with hydrolytic enzyme activity, encoded by chromosomal or plasmid genes, which are referred to as abbreviated “bla”. They are often a part of mobile genetic elements such as transposons or integrons and, therefore, can be easily transferred between bacteria [89]. These genes can be expressed constitutively or in an inducible β-lactam-dependent way. To date, more than 2000 β-lactamases have been identified. There are two main classifications of these large enzyme groups [90]. Classification according to Ambler, which is based on amino acid sequence homology, divides β-lactamases into four classes, named as A, B, C, and D. On the contrary, an updated version of the Busch–Jakoby functional division distinguishes three (originally four) groups of β-lactam enzymes, which are numbered 1 to 3, depending on their substrate preference and inhibitor action profile [91]. Representatives of classes A, C, and D (Ambler classification) as well as members of groups 1 and 3 (Busch–Jakoby classification) are serine-containing enzymes in the active center; therefore, they are called serine-β-lactamases (SBLs). In turn, class B (Ambler classification) and group 3 (Busch–Jakoby classification) include metallo-β-lactamases (MBLs) with a single Zn2+ ion or a pair of Zn2+ ions bound to His/Cys/Asp residues in the active center. The hydrolysis reaction of β-lactams catalyzed by SBL proceeds in two steps. After binding to the antibiotic molecule, the serine within the catalytic center attacks the carbonyl group on the β-lactam ring. This results in the hydrolysis of the amide bond of the β-lactam ring and acylation of the enzyme. Then, with the participation of a water molecule, the enzyme is deacylated and the inactive antibiotic with an open Β-lactam ring is released. A different mechanism is observed for MBL. These enzymes use a zinc cation-coordinated hydroxyl group of the water molecule to inactivate the antibiotic [89][90]. Class A enzymes (subgroups 2a, 2b, 2be, 2br, 2ber, 2c, 2e, and 2f) are the most common of all β-lactamases. Enzymes of this class include PC1 penicillinases encoded by the blaZ gene, showing a narrow spectrum of activity against penicillins, TEM and SHV type β-lactamases hydrolyzing penicillins and early cephalosporins, Klebsiella pneumoniae carbapenemases (KPC) that inactivate carbapenems, as well as extended-spectrum β-lactamases (ESBL), the vast majority of which arise from point mutations altering the hydrolytic preferences of primary TEM (TEM-1, TEM-2) and SHV (SHV-1) [92]. Without counting the enzymes TEM and SHV ESBL, CTX-M, PER, VER, GES, SFO-1, FEC-1, BES-1, BEL-1, TLA-1, and TLA-2 are also found among ESBL [93]. These enzymes have the ability to hydrolyze third-generation cephalosporins known as oxyimino-β-cephalosporins, such as cefotaxime, ceftriaxone, and ceftazidime. Furthermore, they inactivate first- and second-generation cephalosporins and aztreonam, although cephamycins and carbapenems are not their targets. The activity of most representatives of class A β-lactamases, except KPC, is inhibited by clavulanic acid, tazobactam and, to a lesser extent, sulbactam. The class B enzymes (group 3), to which MBLs belong, determine a high level of resistance to penicillins, cephalosporins, and carbapenems, excluding monobactams and aztreonam. Their activity is not inhibited by the previously mentioned β-lactamase inhibitors (clavulanic acid, tazobactam, and sulbactam). However, they are subjected to inhibition by chelating agents such as EDTA, which have a divalent metal ion-binding effect [94]. The substrate spectrum of members of class C (group 1) includes mainly cephalosporins, (except for cefepime, which belongs to the fourth generation cephalosporins), as well as penicillins and monobactams. Like MBLs, β-lactamases of this class do not hydrolyze carbapenems and atreonam (aztreonam). Although they are resistant to β-lactam inhibitors, they can be inhibited by cloxacillin, oxacillin, and aztreonam [95]. Finally, the enzymes of class D (subgroups 2d, 2d, and 2df), to which OXA-type ESBL belong, are characterized by great diversity with regard to their functional properties. As their name suggests, they hydrolyze not only oxacillin but also cloxacillin, carbapenems, penicillins, and to a limited extent, cephalosporins. However, taking into account their inhibitor profile, in most cases their enzymatic activity is not affected by β-lactamase inhibitors [92][96].
Among the novel β-lactamase inhibitors is a relabactam (a diazabicyclooctane beta-lactamase inhibitor), which specifically targets classes A and C of β-lactamases. It was approved by the FDA in 2019 in combination with imipenem (a carbapenem) and cilastatin (a renal dehydropeptidase-I inhibitor) for the treatment of complicated urinary tract infections (UTIs), pyelonephritis, and complicated intra-abdominal infections in adults. The imipenem/cilastatin/relebactam (Recarbrio™) combination exhibits a synergistic effect: (i) imipenem inactivates PBBs and inhibits the cross-linking of peptidoglycan during cell wall synthesis, and its action is protected by (ii) cilastatin, which reduces imipenem renal metabolism, and iii) relebactam, which protects the imipenem from degradation by Ambler classes A and C β-lactamases and Pseudomonas-derived cephalosporinas [97].
Similarly, microbial resistance to macrolides can result from enzymatic inactivation of the antibiotic molecule, which is driven by esterases, such as EreA, EreA2, EreB, EreC, and EreD. These enzymes hydrolyze the macrolide lactone ring. Ere esterases are capable of inactivating macrolides with a 14- and 15-member lactone ring, but not those with a 16-member lactone ring. In addition to EreD, which is chromosomally encoded, all other esterases of the Ere family are encoded by genes located in mobile genetic elements. The occurrence of EreA and EreA2 has been described in many pathogenic clinical strains, including non-typhoidal Salmonella enterica, Pseudomonas spp., Vibrio cholera, and Klebsiella spp. EreB, which is distantly related to these esterases, is the most prevalent isolate among all environmental isolates. In the case of EreC, the ereC gene has been identified in the Enterobacteriaceae genome [98][99]. In contrast, the presence of the ereD gene, which was found in Riemerella anatipestifer isolates from ducks, has not yet been found in other bacterial species [100] (Figure 4(1)).
Figure 4. Representation of the enzymatic inactivation of antibiotics through (1) hydrolysis, (2) group transfer, and (3) the redox process. Figure created with Biorender.com.
The inhibition of the antibacterial activity of macrolides with 14-, 15- and 16-membered lactone rings may also be a consequence of their structure modification, involving phosphorylation of the hydroxyl group located in carbon atom C5 of the antibiotic deosamine moiety. So far, 15 macrolide phosphotransferases have been described: MphA, MphB, MphC, MphD, MphE, MphF, MphG, MphH MphI, MPhJ, MphK, MphL, MphM, MphN, MphO. All of them are encoded by genes located on the chromosome or mobile genetic elements. Their occurrence has been confirmed in many bacterial species, both Gram-positive, e.g., Staphylococcus, and Gram-negative, e.g., Escherichia coli [98].
In addition, the resistance to aminoglycosides depends on phosphotransferase [APH], nucleotidyltransferase [ANT], and acetyltransferase [ACC] activities. The genes encoding these enzymes are located mainly in plasmids, integrons, transposons, or gene cassettes, which together promote their spread throughout bacterial populations [101][102][103][104][105]. Aminoglycoside O-phosphotransferases (APH), which are mainly found among staphylococci and enterococci, catalyze the transfer of a phosphate group from donor ATP (or, in some cases, GTP) to the hydroxyl residue of the aminoglycoside molecule. The APHs are divided into APH(2′), APH(3′), APH(3″), APH(4), APH(6), APH(7″), and APH(9) classes, the most common of which is APH (3′), conferring resistance to kanamycin, neomycin, paromomycin, and others [101][104]. The adenylation of aminoglycosides is another mechanism involved in their inactivation. This, in turn, involves the ATP-dependent transfer of the AMP group to the hydroxyl residue in the aminoglycoside molecule.
The aminoglycoside O-nucleotidyltransferases (ANT) that catalyze this reaction are classified as ANT(2″), ANT(3″), ANT(4′), ANT(6), and ANT(9), the most common of which is ANT(3′′), whose substrate spectrum is limited to streptomycin and spectrinomycin as well as their derivatives. The genes encoding these enzymes have been detected in many Gram-negative bacterial species, including Escherichia coli, Salmonella spp., Pseudomonas aeruginosa, and Klebsiella pneumoniae [101][102]. Aminoglycoside N-acetyltransferases (AACs) are the last group of enzymes that confer resistance to aminoglycosides by acetylation of one of the four amino groups (-NH2) in the antibiotic molecule, using acetyl-coenzyme A as a source of acetyl residues. Enzymes of this type include AAC(1), AAC(2), AAC(3′), and AAC(6′) subclasses. These enzymes determine high levels of resistance to gentamicin in several of both Gram-positive and Gram-negative bacterial species [101][102][106], by promoting changes in the aminoglycoside molecule structure and inhibiting its binding to the target site of action, i.e., 16S rRNA of the 30S ribosome subunit (Figure 4(2)).
The above-mentioned mechanisms of antibiotic inactivation through the redox process underlie the resistance of bacteria, e.g., Sphinogbacterium spp., to tetracyclines. This inactivation involves a flavin monooxygenase that requires molecular oxygen and NADPH for its activity. This enzyme, encoded by the tet(X) gene, catalyzes the hydroxylation of the tetracycline molecule at the position C-11a. The newly formed lla-hydroxytetracycline has a lower magnesium ion coordination capacity than tetracycline and, therefore, does not inhibit protein translation [107][108] (Figure 4(3)).
This entry is adapted from the peer-reviewed paper 10.3390/ijms24065777
References
- Hawkey, P.M. The origins and molecular basis of antibiotic resistance. BMJ 1998, 317, 657–660.
- Hackbarth, C.J.; Kocagoz, T.; Kocagoz, S.; Chambers, H.F. Point mutations in Staphylococcus aureus PBP 2 gene affect penicillin-binding kinetics and are associated with resistance. Antimicrob. Agents Chemother. 1995, 39, 103–106.
- Pinho, M.G.; de Lencastre, H.; Tomasz, A. An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. USA 2001, 98, 10886–10891.
- Raze, D.; Dardenne, O.; Hallut, S.; Martinez-Bueno, M.; Coyette, J.; Ghuysen, J.-M. The Gene Encoding the Low-Affinity Penicillin-Binding Protein 3r in Enterococcus hirae S185R Is Borne on a Plasmid Carrying Other Antibiotic Resistance Determinants. Antimicrob. Agents Chemother. 1998, 42, 534–539.
- Harimaya, A.; Koizumi, J.-I.; Yamazaki, N.; Himi, T.; Yokota, S.-I.; Sato, K.; Fujii, N. Alterations of pbp1a, pbp2b, and pbp2x in Streptococcus pneumoniae isolates from children with otolaryngological infectious disease in the Sapporo district of Japan. J. Infect. Chemother. 2006, 12, 366–371.
- Matsuhashi, M.; Song, M.D.; Ishino, F.; Wachi, M.; Doi, M.; Inoue, M.; Ubukata, K.; Yamashita, N.; Konno, M. Molecular cloning of the gene of a penicillin-binding protein supposed to cause high resistance to beta-lactam antibiotics in Staphylococcus aureus. J. Bacteriol. 1986, 167, 975–980.
- Paterson, G.K.; Harrison, E.M.; Holmes, M.A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2013, 22, 42–47.
- Becker, K.; van Alen, S.; Idelevich, E.A.; Schleimer, N.; Seggewiß, J.; Mellmann, A.; Kaspar, U.; Peters, G. Plasmid-Encoded Transferable mecB-Mediated Methicillin Resistance in Staphylococcus aureus. Emerg. Infect. Dis. 2018, 24, 242–248.
- Schwendener, S.; Cotting, K.; Perreten, V. Novel methicillin resistance gene mecD in clinical Macrococcus caseolyticus strains from bovine and canine sources. Sci. Rep. 2017, 7, srep43797.
- Cooper, M.; Fiorini, M.T.; Abell, C.; Williams, D.H. Binding of vancomycin group antibiotics to d -alanine and d -lactate presenting self-assembled monolayers. Bioorganic Med. Chem. 2000, 8, 2609–2616.
- Reynolds, P.E.; Courvalin, P. Vancomycin Resistance in Enterococci Due to Synthesis of Precursors Terminating in d -Alanyl- d -Serine. Antimicrob. Agents Chemother. 2005, 49, 21–25.
- Ge, M.; Chen, Z.; Russell, H.; Onishi, H.R.; Kohler, J.; Silver, L.L.; Kerns, R.; Fukuzawa, S.; Thompson, C.; Kahne, D. Vancomycin Derivatives That Inhibit Peptidoglycan Biosynthesis Without Binding d -Ala- d -Ala. Science 1999, 284, 507–511.
- Ahmed, M.O.; Baptiste, K.E. Vancomycin-Resistant Enterococci: A Review of Antimicrobial Resistance Mechanisms and Perspectives of Human and Animal Health. Microb. Drug Resist. 2018, 24, 590–606.
- Rice, L.B. Mechanisms of Resistance and Clinical Relevance of Resistance to β-Lactams, Glycopeptides, and Fluoroquinolones. Mayo Clin. Proc. 2012, 87, 198–208.
- Gagnon, S.; Lévesque, S.; Lefebvre, B.; Bourgault, A.-M.; Labbé, A.-C.; Roger, M. vanA-containing Enterococcus faecium susceptible to vancomycin and teicoplanin because of major nucleotide deletions in Tn1546. J. Antimicrob. Chemother. 2011, 66, 2758–2762.
- Hill, C.M.; Krause, K.M.; Lewis, S.R.; Blais, J.; Benton, B.M.; Mammen, M.; Humphrey, P.P.; Kinana, A.; Janc, J.W. Specificity of Induction of the vanA and vanB Operons in Vancomycin-Resistant Enterococci by Telavancin. Antimicrob. Agents Chemother. 2010, 54, 2814–2818.
- Moffatt, J.H.; Harper, M.; Boyce, J.D. Mechanisms of Polymyxin Resistance. In Polymyxin Antibiotics: From Laboratory Bench to Bedside; Springer: Berlin/Heidelberg, Germany, 2019; pp. 55–71.
- Hamad, M.A.; Di Lorenzo, F.; Molinaro, A.; Valvano, M.A. Aminoarabinose is essential for lipopolysaccharide export and intrinsic antimicrobial peptide resistance in Burkholderia cenocepacia. Mol. Microbiol. 2012, 85, 962–974.
- Stock, I. Natural Antibiotic Susceptibility of Proteus spp., with Special Reference to P. Mirabilis and P. penneri Strains. J. Chemother. 2003, 15, 12–26.
- Hase, S.; Hofstad, T.; Rietschel, E.T. Chemical structure of the lipid A component of lipopolysaccharides from Fusobacterium nucleatum. J. Bacteriol. 1977, 129, 9–14.
- Johnson, L.; Horsman, S.R.; Charron-Mazenod, L.; Turnbull, A.L.; Mulcahy, H.; Surette, M.G.; Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2013, 13, 115.
- Cheng, H.-Y.; Chen, Y.-F.; Peng, H.-L. Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J. Biomed. Sci. 2010, 17, 60.
- Francis, V.I.; Stevenson, E.C.; Porter, S.L. Two-component systems required for virulence in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2017, 364, fnx104.
- Huang, J.; Li, C.; Song, J.; Velkov, T.; Wang, L.; Zhu, Y.; Li, J. Regulating polymyxin resistance in Gram-negative bacteria: Roles of two-component systems PhoPQ and PmrAB. Futur. Microbiol. 2020, 15, 445–459.
- Marceau, M.; Sebbane, F.; Ewann, F.; Collyn, F.; Lindner, B.; Campos, M.A.; Bengoechea, J.-A.; Simonet, M. The pmrF polymyxin-resistance operon of Yersinia pseudotuberculosis is upregulated by the PhoP-PhoQ two-component system but not by PmrA-PmrB, and is not required for virulence. Microbiology 2004, 150, 3947–3957.
- Gooderham, W.J.; Hancock, R.E.W. Regulation of virulence and antibiotic resistance by two-component regulatory systems in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 2009, 33, 279–294.
- Olaitan, A.; Morand, S.; Rolain, J.-M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643.
- Moffatt, J.H.; Harper, M.; Harrison, P.; Hale, J.D.; Vinogradov, E.; Seemann, T.; Henry, R.; Crane, B.; St. Michael, F.; Cox, A.D.; et al. Colistin Resistance in Acinetobacter baumannii Is Mediated by Complete Loss of Lipopolysaccharide Production. Antimicrob. Agents Chemother. 2010, 54, 4971–4977.
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12–31.
- Jacoby, G.A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 2005, 41, S120–S126.
- Hooper, D.C.; Jacoby, G.A. Topoisomerase Inhibitors: Fluoroquinolone Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025320.
- Ruiz, J. Mechanisms of resistance to quinolones: Target alterations, decreased accumulation and DNA gyrase protection. J. Antimicrob. Chemother. 2003, 51, 1109–1117.
- Wasyl, D.; Hoszowski, A.; Zając, M. Prevalence and characterisation of quinolone resistance mechanisms in Salmonella spp. Vet. Microbiol. 2014, 171, 307–314.
- Eliopoulos, G.M. Quinolone Resistance Mechanisms in Pneumococci. Clin. Infect. Dis. 2004, 38, S346–S349.
- Schmitz, F.J.; Hofmann, B.; Hansen, B.; Scheuring, S.; Lückefahr, M.; Klootwijk, M.; Verhoef, J.; Fluit, A.; Heinz, H.P.; Köhrer, K.; et al. Relationship between ciprofloxacin, ofloxacin, levofloxacin, sparfloxacin and moxifloxacin (BAY 12-8039) MICs and mutations in grlA, grlB, gyrA and gyrB in 116 unrelated clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 1998, 41, 481–484.
- Khan, A.S.; Phelan, J.E.; Khan, M.T.; Ali, S.; Qasim, M.; Napier, G.; Campino, S.; Ahmad, S.; Cabral-Marques, O.; Zhang, S.; et al. Characterization of rifampicin-resistant Mycobacterium tuberculosis in Khyber Pakhtunkhwa, Pakistan. Sci. Rep. 2021, 11, 14194.
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912.
- Koch, A.; Mizrahi, V.; Warner, D.F. The impact of drug resistance on Mycobacterium tuberculosis physiology: What can we learn from rifampicin? Emerg. Microbes Infect. 2014, 3, 1–11.
- Zaw, M.T.; Emran, N.A.; Lin, Z. Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis. J. Infect. Public Health 2018, 11, 605–610.
- Padayachee, T.; Klugman, K.P. Molecular Basis of Rifampin Resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 1999, 43, 2361–2365.
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630.
- Lin, Y.-H.; Tai, C.-H.; Li, C.-R.; Lin, C.-F.; Shi, Z.-Y. Resistance profiles and rpoB gene mutations of Mycobacterium tuberculosis isolates in Taiwan. J. Microbiol. Immunol. Infect. 2013, 46, 266–270.
- Weisblum, B. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 1995, 39, 577–585.
- Schroeder, M.R.; Stephens, D.S. Macrolide Resistance in Streptococcus pneumoniae. Front. Cell. Infect. Microbiol. 2016, 6, 98.
- Leclercq, R. Mechanisms of Resistance to Macrolides and Lincosamides: Nature of the Resistance Elements and Their Clinical Implications. Clin. Infect. Dis. 2002, 34, 482–492.
- Daoud, Z.; Kourani, M.; Saab, R.; Nader, M.A.; Hajjar, M. Resistance of Streptococcus pneumoniae isolated from Lebanese patients between 2005 and 2009. Rev. Esp. Quimioter. Publ. Soc. Esp. Quimioter. 2011, 24, 84–90.
- Portillo, A.; Ruiz-Larrea, F.; Zarazaga, M.; Alonso, A.; Martinez, J.L.; Torres, C. Macrolide Resistance Genes in Enterococcus spp. Antimicrob. Agents Chemother. 2000, 44, 967–971.
- Westh, H.; Hougaard, D.M.; Vuust, J.; Rosdahl, V.T. erm genes in erythromycin-resistant Staphylococcus aureus and coagulase-negative staphylococci. APMIS Acta Pathol. Microbiol. Immunol. Scand. 1995, 103, 225–232.
- De Leener, E.; Martel, A.; Decostere, A.; Haesebrouck, F. Distribution of the erm(B) Gene, tet racycline Resistance Genes, and Tn 1545-like Transposons in Macrolide- and Lincosamide-Resistant Enterococci from Pigs and Humans. Microb. Drug Resist. 2004, 10, 341–345.
- Huber, L.; Giguère, S.; Slovis, N.M.; Álvarez-Narváez, S.; Hart, K.A.; Greiter, M.; Morris, E.R.A.; Cohen, N.D. The novel and transferable erm (51) gene confers macrolides, lincosamides and streptogramins B (MLSB) resistance to clonal Rhodococcus equi in the environment. Environ. Microbiol. 2020, 22, 2858–2869.
- Wipf, J.R.K.; Schwendener, S.; Perreten, V. The Novel Macrolide-Lincosamide-Streptogramin B Resistance Gene erm (44) Is Associated with a Prophage in Staphylococcus xylosus. Antimicrob. Agents Chemother. 2014, 58, 6133–6138.
- Chen, H.; Wang, X.; Yin, Y.; Li, S.; Zhang, Y.; Wang, Q.; Wang, H. Molecular characteristics of oxazolidinone resistance in enterococci from a multicenter study in China. BMC Microbiol. 2019, 19, 162.
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA Methyltransferase Confers Resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A Antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505.
- Doi, Y.; Wachino, J.-I.; Arakawa, Y. Aminoglycoside Resistance. Infect. Dis. Clin. N. Am. 2016, 30, 523–537.
- Zaman, S.; Fitzpatrick, M.; Lindahl, L.; Zengel, J. Novel mutations in ribosomal proteins L4 and L22 that confer erythromycin resistance in Escherichia coli. Mol. Microbiol. 2007, 66, 1039–1050.
- Bilgin, N.; Richter, A.A.; Ehrenberg, M.; Dahlberg, A.E.; Kurland, C.G. Ribosomal RNA and protein mutants resistant to spectinomycin. EMBO J. 1990, 9, 735–739.
- Kondo, J. A Structural Basis for the Antibiotic Resistance Conferred by an A1408G Mutation in 16S rRNA and for the Antiprotozoal Activity of Aminoglycosides. Angew. Chem. Int. Ed. 2011, 51, 465–468.
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. MedChemComm 2015, 7, 11–27.
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 808–816.
- Yarlagadda, V.; Manjunath, G.B.; Sarkar, P.; Akkapeddi, P.; Paramanandham, K.; Shome, B.R.; Ravikumar, R.; Haldar, J. Glycopeptide Antibiotic To Overcome the Intrinsic Resistance of Gram-Negative Bacteria. ACS Infect. Dis. 2015, 2, 132–139.
- Konovalova, A.; Kahne, D.E.; Silhavy, T.J. Outer Membrane Biogenesis. Annu. Rev. Microbiol. 2017, 71, 539–556.
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960.
- Pagès, J.-M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903.
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656.
- Kumar, A.; Schweizer, H.P. Bacterial resistance to antibiotics: Active efflux and reduced uptake. Adv. Drug Deliv. Rev. 2005, 57, 1486–1513.
- Khalid, A.; Lubián, A.F.; Ma, L.; Lin, R.C.; Iredell, J.R. Characterizing the role of porin mutations in susceptibility of beta lactamase producing Klebsiella pneumoniae isolates to ceftaroline and ceftaroline-avibactam. Int. J. Infect. Dis. 2020, 93, 252–257.
- Doménech-Sánchez, A.; Hernández-Allés, S.; Martínez-Martínez, L.; Benedí, V.J.; Albertí, S. Identification and Characterization of a New Porin Gene of Klebsiella pneumoniae: Its Role in β-Lactam Antibiotic Resistance. J. Bacteriol. 1999, 181, 2726–2732.
- Khalifa, S.M.; El-Aziz, A.M.A.; Hassan, R.; Abdelmegeed, E.S. β-lactam resistance associated with β-lactamase production and porin alteration in clinical isolates of E. coli and K. pneumoniae. PLoS ONE 2021, 16, e0251594.
- El Amin, N.; Giske, C.G.; Jalal, S.; Keijser, B.; Kronvall, G.; Wretlind, B. Carbapenem resistance mechanisms in Pseudomonas aeruginosa: Alterations of porin OprD and efflux proteins do not fully explain resistance patterns observed in clinical isolates. Apmis 2005, 113, 187–196.
- Studemeister, A.E.; Quinn, J.P. Selective imipenem resistance in Pseudomonas aeruginosa associated with diminished outer membrane permeability. Antimicrob. Agents Chemother. 1988, 32, 1267–1268.
- Takada, H.; Yoshikawa, H. Essentiality and function of WalK/WalR two-component system: The past, present, and future of research. Biosci. Biotechnol. Biochem. 2018, 82, 741–751.
- Peng, H.; Hu, Q.; Shang, W.; Yuan, J.; Zhang, X.; Liu, H.; Zheng, Y.; Hu, Z.; Yang, Y.; Tan, L.; et al. WalK(S221P), a naturally occurring mutation, confers vancomycin resistance in VISA strain XN108. J. Antimicrob. Chemother. 2016, 72, 1006–1013.
- Howden, B.P.; McEvoy, C.R.E.; Allen, D.L.; Chua, K.; Gao, W.; Harrison, P.; Bell, J.; Coombs, G.; Bennett-Wood, V.; Porter, J.L.; et al. Evolution of Multidrug Resistance during Staphylococcus aureus Infection Involves Mutation of the Essential Two Component Regulator WalKR. PLoS Pathog. 2011, 7, e1002359.
- Fernández, L.; Hancock, R.E.W. Adaptive and Mutational Resistance: Role of Porins and Efflux Pumps in Drug Resistance. Clin. Microbiol. Rev. 2013, 26, 163.
- Kornelsen, V.; Kumar, A. Update on Multidrug Resistance Efflux Pumps in Acinetobacter spp. Antimicrob. Agents Chemother. 2021, 65, e00514-21.
- Aron, Z.; Opperman, T.J. The hydrophobic trap-the Achilles heel of RND efflux pumps. Res. Microbiol. 2018, 169, 393–400.
- Srinivasan, V.B.; Rajamohan, G.; Gebreyes, W.A. Role of AbeS, a Novel Efflux Pump of the SMR Family of Transporters, in Resistance to Antimicrobial Agents in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2009, 53, 5312–5316.
- Hassan, K.A.; Liu, Q.; Henderson, P.J.F.; Paulsen, I.T. Homologs of the Acinetobacter baumannii AceI Transporter Represent a New Family of Bacterial Multidrug Efflux Systems. Mbio 2015, 6, e01982-14.
- Bolla, J.R.; Howes, A.C.; Fiorentino, F.; Robinson, C.V. Assembly and regulation of the chlorhexidine-specific efflux pump AceI. Proc. Natl. Acad. Sci. USA 2020, 117, 17011–17018.
- Kabra, R.; Chauhan, N.; Kumar, A.; Ingale, P.; Singh, S. Efflux pumps and antimicrobial resistance: Paradoxical components in systems genomics. Prog. Biophys. Mol. Biol. 2018, 141, 15–24.
- Rouquette-Loughlin, C.; Dunham, S.A.; Kuhn, M.; Balthazar, J.T.; Shafer, W.M. The NorM Efflux Pump of Neisseria gonorrhoeae and Neisseria meningitidis Recognizes Antimicrobial Cationic Compounds. J. Bacteriol. 2003, 185, 1101–1106.
- Hürlimann, L.M.; Hohl, M.; Seeger, M.A. Split tasks of asymmetric nucleotide-binding sites in the heterodimeric ABC exporter EfrCD. FEBS J. 2017, 284, 1672–1687.
- Greene, N.P.; Kaplan, E.; Crow, A.; Koronakis, V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front. Microbiol. 2018, 9, 950.
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418.
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501.
- Goli, H.R.; Nahaei, M.R.; Rezaee, M.A.; Hasani, A.; Kafil, H.S.; Aghazadeh, M.; Nikbakht, M.; Khalili, Y. Role of MexAB-OprM and MexXY-OprM efflux pumps and class 1 integrons in resistance to antibiotics in burn and Intensive Care Unit isolates of Pseudomonas aeruginosa. J. Infect. Public Health 2018, 11, 364–372.
- Beggs, G.A.; Brennan, R.G.; Arshad, M. MarR family proteins are important regulators of clinically relevant antibiotic resistance. Protein Sci. 2019, 29, 647–653.
- Varela, M.; Kumar, S. Molecular mechanisms of bacterial resistance to antimicrobial agents. Chemotherapy 2013, 14, 522–534.
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201.
- Bonomo, R.A. β-Lactamases: A Focus on Current Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a025239.
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976.
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J. Intensiv. Care 2020, 8, 13.
- Bradford, P.A. Extended-Spectrum β-Lactamases in the 21st Century: Characterization, Epidemiology, and Detection of This Important Resistance Threat. Clin. Microbiol. Rev. 2001, 14, 933–951.
- Yong, D.; Lee, K.; Yum, J.H.; Shin, H.B.; Rossolini, G.M.; Chong, Y. Imipenem-EDTA Disk Method for Differentiation of Metallo-β-Lactamase-Producing Clinical Isolates of Pseudomonas spp. and Acinetobacter spp. J. Clin. Microbiol. 2002, 40, 3798–3801.
- Jacoby, G.A. AmpC β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182.
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233.
- Heo, Y.A. Imipenem/Cilastatin/Relebactam: A Review in Gram-Negative Bacterial Infections. Drugs 2021, 81, 377–388.
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and Outlook on Enzyme-Mediated Macrolide Resistance. Front. Microbiol. 2018, 9, 1942.
- Zieliński, M.; Park, J.; Sleno, B.; Berghuis, A.M. Structural and functional insights into esterase-mediated macrolide resistance. Nat. Commun. 2021, 12, 1732.
- Xing, L.; Yu, H.; Qi, J.; Jiang, P.; Sun, B.; Cui, J.; Ou, C.; Chang, W.; Hu, Q. ErmF and ereD Are Responsible for Erythromycin Resistance in Riemerella anatipestifer. PLoS ONE 2015, 10, e0131078.
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171.
- Jaimee, G.; Halami, P.M. Emerging resistance to aminoglycosides in lactic acid bacteria of food origin—An impending menace. Appl. Microbiol. Biotechnol. 2015, 100, 1137–1151.
- Disney, M.D. Studying Modification of Aminoglycoside Antibiotics by Resistance-Causing Enzymes via Microarray. Breast Cancer 2011, 808, 303–320.
- Jana, S.; Deb, J.K. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 2006, 70, 140–150.
- Labby, K.J.; Garneau-Tsodikova, S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Futur. Med. Chem. 2013, 5, 1285–1309.
- Gonzalez, L.S.; Spencer, J.P. Aminoglycosides: A practical review. Am. Fam. Physician 1998, 58, 1811.
- Yang, W.; Moore, I.F.; Koteva, K.P.; Bareich, D.C.; Hughes, D.W.; Wright, G.D. TetX Is a Flavin-dependent Monooxygenase Conferring Resistance to Tetracycline Antibiotics. J. Biol. Chem. 2004, 279, 52346–52352.
- Markley, J.L.; Wencewicz, T.A. Tetracycline-Inactivating Enzymes. Front. Microbiol. 2018, 9, 1058.
This entry is offline, you can click here to edit this entry!