Pathologically, Parkinson’s disease (PD) is characterized by the loss of dopamine (DA) neurons in the substantia nigra pars compacta (SNc) and the presence of intracellular inclusions, called Lewy bodies and Lewy neurites, composed mostly of α-synuclein (α-Syn). Much of PD research has focused on the role of α-Syn aggregates in the degeneration of SNc DA neurons due to the impact of striatal DA deficits on classical motor phenotypes. However, abundant Lewy pathology is also found in other brain regions including the midbrain raphe nuclei, which may contribute to non-motor symptoms. Indeed, dysfunction of the serotonergic (5-HT) system, which regulates mood and emotional pathways, occurs during the premotor phase of PD.
- depression
- Parkinson’s disease
- α-synuclein
- serotonin
- raphe nuclei
1. Introduction
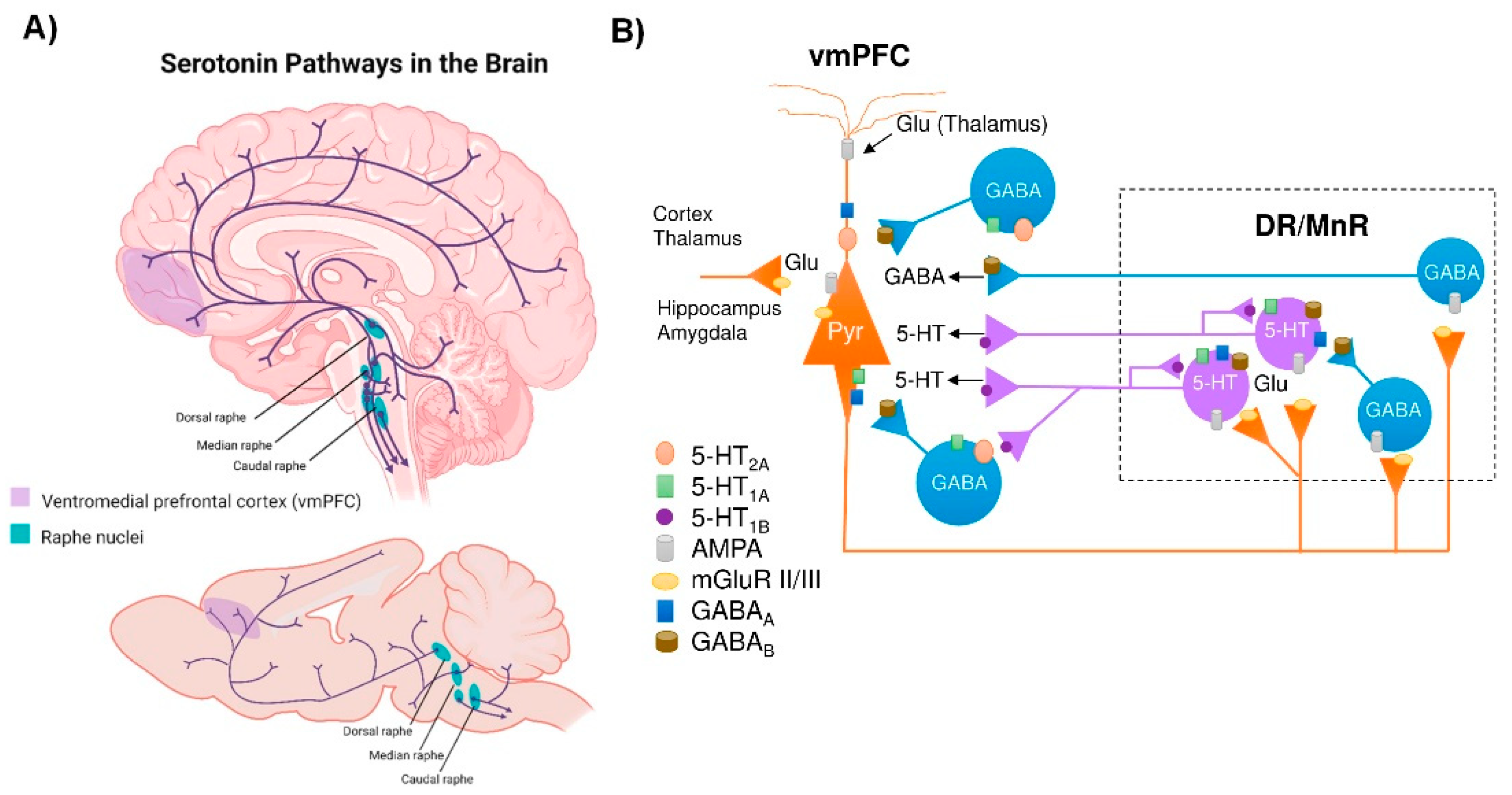
2. Connectivity of the Brain Serotonin System
3. α-Synuclein and Serotonin Neurotransmission
4. Dysfunction of the 5-HT System in PD Patients
5. Dysfunction of the 5-HT System in Animal Models with Overexpression of α-Syn
6. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11020541
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376.
- Lima, M.; Martins, E.; Delattre, A.; Proenc, M.; Mori, M.; Carabelli, B. Motor and nonmotor features of Parkinson’s disease: A review of clinical and experimental studies. CNS Neurol. Disord. Drug Targets 2012, 11, 439–449.
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601.
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of both dopaminergic and serotonergic axons—A common mechanism in Parkinson’s disease. Front. Cell. Neurosci. 2016, 10, 293.
- Maillet, A.; Krack, P.; Lhommée, E.; Météreau, E.; Klinger, H.; Favre, E.; Le Bars, D.; Schmitt, E.; Bichon, A.; Pelissier, P.; et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain 2019, 139, 2486–2502.
- Aarsland, D.; Marsh, L.; Schrag, A. Neuropsychiatric symptoms in Parkinson’s disease. Mov. Disord. 2009, 24, 2175–2186.
- Santangelo, G.; Vitale, C.; Trojano, L.; Longo, K.; Cozzolino, A.; Grossi, D.; Barone, P. Relationship between depression and cognitive dysfunctions in Parkinson’s disease without dementia. J. Neurol. 2009, 256, 632–638.
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474.
- Thobois, S.; Ardouin, C.; Lhommée, E.; Klinger, H.; Lagrange, C.; Xie, J.; Fraix, V.; Coelho Braga, M.C.; Hassani, R.; Kistner, A.; et al. Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: Predictors and underlying mesolimbic denervation. Brain 2010, 133, 1111–1127.
- De la Riva, P.; Smith, K.; Xie, S.X.; Weintraub, D. Course of psychiatric symptoms and global cognition in early Parkinson disease. Neurology 2014, 83, 1096–1103.
- Dujardin, K.; Langlois, C.; Plomhause, L.; Carette, A.S.; Delliaux, M.; Duhamel, A.; Defebvre, L. Apathy in untreated early-stage Parkinson disease: Relationship with other non-motor symptoms. Mov. Disord 2014, 29, 1796–1801.
- Schrag, A.; Sauerbier, A.; Chaudhuri, K.R. New clinical trials for nonmotor manifestations of Parkinson’s disease. Mov. Disord 2015, 30, 1490–1504.
- Santos García, D.; de Deus Fonticoba, T.; Suárez Castro, E.; Borrué, C.; Mata, M.; Solano Vila, B.; Cots Foraster, A.; Álvarez Sauco, M.; Rodríguez Pérez, A.B.; Vela, L.; et al. Non-motor symptoms burden, mood, and gait problems are the most significant factors contributing to a poor quality of life in non-demented Parkinson’s disease patients: Results from the COPPADIS Study Cohort. Park. Relat. Disord. 2019, 66, 151–157.
- Reijnders, J.S.; Ehrt, U.; Weber, W.E.; Aarsland, D.; Leentjens, F. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov. Disord. 2008, 23, 183–189.
- Aarsland, D.; Påhlhagen, S.; Ballard, C.G.; Ehrt, U.; Svenningsson, P. Depression in Parkinson disease--epidemiology, mechanisms and management. Nat. Rev. Neurol. 2011, 8, 35–47.
- Chuquilín-Arista, F.; Álvarez-Avellón, T.; Menéndez-González, M. Prevalence of Depression and Anxiety in Parkinson Disease and Impact on Quality of Life: A Community-Based Study in Spain. J. Geriatr. Psychiatry Neurol. 2020, 33, 207–213.
- Karlsen, K.H.; Tandberg, E.; Aarsland, D.; Larsen, J.P. Health related quality of life in Parkinson’s disease: A prospective longitudinal study. J. Neurol. Neurosurg. Psychiatry 2000, 69, 584–589.
- Den Oudsten, B.; Van Heck, G.; De Vries, J. Quality of life and related concepts in Parkinson’s disease: A systematic review. Mov. Disord. 2007, 22, 1528–1537.
- Soh, S.; Morris, M.; McGinley, J. Determinants of health-related quality of life in Parkinson’s disease: A systematic review. Park. Relat. Disord. 2011, 17, 1–9.
- Hemmerle, A.; Herman, J.; Seroog, K. Stress, depression and Parkinson’s disease. Exp. Neurol. 2012, 233, 79–86.
- Greffard, S.; Verny, M.; Bonnet, A.M.; Beinis, J.Y.; Gallinari, C.; Meaume, S.; Piette, F.; Hauw, J.J.; Duyckaerts, C. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch. Neurol. 2006, 63, 584–588.
- Halliday, G.; Lees, A.; Stern, M. Milestones in Parkinson’s disease—Clinical and pathologic features. Mov. Disord. 2011, 26, 1015–1021.
- Hornykiewicz, O. 50 years of levodopa. Mov. Disord. 2015, 30, 1008.
- Foffani, G.; Obeso, J.A. A Cortical Pathogenic Theory of Parkinson’s Disease. Neuron 2018, 99, 1116–1128.
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056.
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134.
- Braak, H.; Del Tredici, K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: Separating the wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157.
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of alpha-synuclein. Cell Tissue Res. 2018, 373, 137–148.
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668.
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109.
- Brichta, L.; Greengard, P.; Flajolet, M. Advances in the pharmacological treatment of Parkinson’s disease: Targeting neurotransmitter systems. Trends Neurosci. 2013, 36, 543–554.
- Giguère, N.; Burke Nanni, S.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455.
- Van Den Berge, N.; Ulusoy, A. Animal models of brain-first and body-first Parkinson’s disease. Neurobiol. Dis. 2022, 163, 105599.
- Harding, A.J.; Stimson, E.; Henderson, J.M.; Halliday, G.M. Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain 2002, 125, 2431–2445.
- Stoyka, L.E.; Arrant, A.E.; Thrasher, D.R.; Russell, D.L.; Freire, J.; Mahoney, C.L.; Narayanan, A.; Dib, A.G.; Standaert, D.G.; Volpicelli-Daley, L.A. Behavioral defects associated with amygdala and cortical dysfunction in mice with seeded α-synuclein inclusions. Neurobiol. Dis. 2020, 134, 104708.
- Hornung, J.P. The human raphe nuclei and the serotonergic system. J. Chem. Neuroanat. 2003, 26, 331–343.
- Descarries, L.; Riad, M.; Parent, M. Ultrastructure of the Serotonin Innervation in the Mammalian Central Nervous System. In Handbook of Behavioral Neurobiology of Serotonin; Müller, C.P., Jacobs, B.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2010.
- Sparta, D.R.; Stuber, G.D. Cartography of serotonergic circuits. Neuron 2014, 83, 513–515.
- Bockaert, J.; Claeysen, S.A.D.; Marin, P. Classification and Signaling Characteristics of 5-HT Receptors. In Handbook of Behavioral Neurobiology of Serotonin; Müller, C.P., Jacobs, B.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2010.
- Weissbourd, B.; Ren, J.; DeLoach, K.E.; Guenthner, C.J.; Miyamichi, K.; Luo, L. Presynaptic partners of dorsal raphe serotonergic and GABAergic neurons. Neuron 2014, 83, 645–662.
- Mengod, G.; Palacios, J.M.; Cortés, R. Cartography of 5-HT1A and 5-HT2A Receptor Subtypes in Prefrontal Cortex and Its Projections. ACS Chem. Neurosci. 2015, 6, 1089–1098.
- Amargós-Bosch, M.; Bortolozzi, A.; Puig, M.V.; Serrats, J.; Adell, A.; Celada, P.; Toth, M.; Mengod, G.; Artigas, F. Co-expression and in vivo interaction of serotonin1A and serotonin2A receptors in pyramidal neurons of prefrontal cortex. Cereb. Cortex 2004, 14, 281–299.
- Santana, N.; Bortolozzi, A.; Serrats, J.; Mengod, G.; Artigas, F. Expression of serotonin1A and serotonin2A receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb. Cortex 2004, 14, 1100–1109.
- López-Terrones, E.; Celada, P.; Riga, M.S.; Artigas, F. Preferential In Vivo Inhibitory Action of Serotonin in Rat Infralimbic versus Prelimbic Cortex: Relevance for Antidepressant Treatments. Cereb. Cortex 2022, 32, 3000–3013.
- Janušonis, S. Serotonin in space: Understanding single fibers. ACS Chem. Neurosci. 2017, 8, 893–896.
- Gabbott, P.L.; Warner, T.A.; Jays, P.R.; Salway, P.; Busby, S.J. Prefrontal cortex in the rat: Projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol. 2005, 492, 145–177.
- Muzerelle, A.; Scotto-Lomassese, S.; Bernard, J.F.; Soiza-Reilly, M.; Gaspar, P. Conditional anterograde tracing reveals distinct targeting of individual serotonin cell groups (B5-B9) to the forebrain and brainstem. Brain Struct. Funct. 2016, 221, 535–561.
- George, J.M. The synucleins. Genome Biol. 2002, 3, 1–6.
- Li, J.Y.; Henning Jensen, P.; Dahlström, A. Differential localization of α-, β-and γ-synucleins in the rat CNS. Neuroscience 2002, 113, 463–478.
- Longhena, F.; Faustini, G.; Spillantini, M.G.; Bellucci, A. Living in Promiscuity: The Multiple Partners of Alpha-Synuclein at the Synapse in Physiology and Pathology. Int. J. Mol. Sci. 2019, 20, 141.
- Pavia-Collado, R.; Rodríguez-Aller, R.; Alarcón-Arís, D.; Miquel-Rio, L.; Ruiz-Bronchal, E.; Paz, V.; Campa, L.; Galofré, M.; Sgambato, V.; Bortolozzi, A. Up and Down γ-Synuclein Transcription in Dopamine Neurons Translates into Changes in Dopamine Neurotransmission and Behavioral Performance in Mice. Int. J. Mol. Sci. 2022, 23, 1807.
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69.
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073.
- Deleersnijder, A.; Gerard, M.; Debyser, Z.; Baekelandt, V. The remarkable conformational plasticity of alpha-synuclein: Blessing or curse? Trends Mol. Med. 2013, 19, 368–377.
- Lv, Z.; Krasnoslobodtsev, A.V.; Zhang, Y.; Ysselstein, D.; Rochet, J.C.; Blanchard, S.C.; Lyubchenko, Y.L. Effect of acidic pH on the stability of alpha-synuclein dimers. Biopolymers 2016, 105, 715–724.
- Galvagnion, C. The Role of Lipids Interacting with alpha-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Park. Dis. 2017, 7, 433–450.
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486.
- Vidović, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732.
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815.
- Withers, G.S.; George, J.M.; Banker, G.A.; Clayton, D.F. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 1997, 99, 87–94.
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van Der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373.
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252.
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79.
- Burre, J. The Synaptic Function of alpha-Synuclein. J. Parkinsons Dis. 2015, 5, 699–713.
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091.
- Fernández-Nogales, M.; López-Cascales, M.T.; Murcia-Belmonte, V.; Escalante, A.; Fernández-Albert, J.; Muñoz-Viana, R.; Barco, A.; Herrera, E. Multiomic Analysis of Neurons with Divergent Projection Patterns Identifies Novel Regulators of Axon Pathfinding. Adv Sci. 2022, 9, e2200615.
- Sidhu, A.; Wersinger, C.; Vernier, P. Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J. 2004, 18, 637–647.
- Wersinger, C.; Jeannotte, A.; Sidhu, A. Attenuation of the norepinephrine transporter activity and trafficking via interactions with alpha-synuclein. Eur. J. Neurosci. 2006, 24, 3141–3152.
- Wersinger, C.; Rusnak, M.; Sidhu, A. Modulation of the trafficking of the human serotonin transporter by human alpha-synuclein. Eur. J. Neurosci. 2006, 24, 55–64.
- Oaks, A.W.; Sidhu, A. Synuclein modulation of monoamine transporters. FEBS Lett. 2011, 585, 1001–1006.
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by alpha-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554.
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol. Ther. 2018, 26, 550–567.
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma membrane monoamine transporters: Structure, regulation and function. Nat. Rev. Neurosci. 2003, 4, 13–25.
- Hahn, M.K.; Blakely, R.D. Monoamine transporter gene structure and polymorphisms in relation to psychiatric and other complex disorders. Pharm. J. 2002, 2, 217–235.
- Wersinger, C.; Sidhu, A. Partial regulation of serotonin transporter function by gamma-synuclein. Neurosci. Lett. 2009, 453, 157–161.
- Miquel-Rio, L.; Alarcón-Arís, D.; Torres-López, M.; Cóppola-Segovia, V.; Pavia-Collado, R.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Casal, C.; Montefeltro, A.; et al. Human α-synuclein overexpression in mouse serotonin neurons triggers a depressive-like phenotype. Rescue by oligonucleotide therapy. Transl. Psychiatry 2022, 12, 79.
- Narboux-Nême, N.; Sagné, C.; Doly, S.; Diaz, S.L.; Martin, C.B.; Angenard, G.; Martres, M.P.; Giros, B.; Hamon, M.; Lanfumey, L.; et al. Severe serotonin depletion after conditional deletion of the vesicular monoamine transporter 2 gene in serotonin neurons: Neural and behavioral consequences. Neuropsychopharmacology 2011, 36, 2538–2550.
- Eiden, L.E.; Weihe, E. VMAT2: A dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse. Ann. N. Y. Acad. Sci. 2011, 1216, 86–98.
- Yamamoto, S.; Fukae, J.; Mori, H.; Mizuno, Y.; Hattori, N. Positive immunoreactivity for vesicular monoamine transporter 2 in Lewy bodies and Lewy neurites in substantia nigra. Neurosci. Lett. 2006, 396, 187–191.
- Taylor, T.N.; Caudle, W.M.; Shepherd, K.R.; Noorian, A.; Jackson, C.R.; Iuvone, P.M.; Weinshenker, D.; Greene, J.G.; Miller, G.W. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J. Neurosci. 2009, 29, 8103–8113.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- De Pablo-Fernández, E.; Lees, A.J.; Holton, J.L.; Warner, T.T. Neuropathological progression of clinical Parkinson disease subtypes. Nat. Rev. Neurol. 2019, 15, 361.
- Iranzo, A.; Tolosa, E.; Gelpi, E.; Molinuevo, J.L.; Valldeoriola, F.; Serradell, M.; Sanchez-Valle, R.; Vilaseca, I.; Lomeña, F.; Vilas, D.; et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: An observational cohort study. Lancet Neurol. 2013, 12, 443–453.
- Boileau, I.; Warsh, J.J.; Guttman, M.; Saint-Cyr, J.A.; McCluskey, T.; Rusjan, P.; Houle, S.; Wilson, A.A.; Meyer, J.H.; Kish, S.J. Elevated serotonin transporter binding in depressed patients with Parkinson’s disease: A preliminary PET study with DASB. Mov. Disord. 2008, 23, 1776–1780.
- Politis, M.; Wu, K.; Loane, C.; Turkheimer, F.E.; Molloy, S.; Brooks, D.J.; Piccini, P. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology 2010, 75, 1920–1927.
- Ballanger, B.; Klinger, H.; Eche, J.; Lerond, J.; Vallet, A.E.; Le Bars, D.; Tremblay, L.; Sgambato-Faure, V.; Broussolle, E.; Thobois, S. Role of serotonergic 1A receptor dysfunction in depression associated with Parkinson’s disease. Mov. Disord. 2012, 27, 84–89.
- Ohno, Y.; Shimizu, S.; Tokudome, K.; Kunisawa, N.; Sasa, M. New insights into the therapeutic role of the serotonergic system in Parkinson’s disease. Prog. Neurobiol. 2015, 134, 104–121.
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Koros, C.; Yousaf, T.; Picillo, M.; Polychronis, S.; Simitsi, A.; Giordano, B.; Chappell, Z.; et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: A cross-sectional study. Lancet Neurol. 2019, 18, 748–759.
- Tan, S.K.; Hartung, H.; Sharp, T.; Temel, Y. Serotonin-dependent depression in Parkinson’s disease: A role for the subthalamic nucleus? Neuropharmacology 2011, 61, 387–399.
- Haapaniemi, T.H.; Ahonen, A.; Torniainen, P.; Sotaniemi, K.A.; Myllylä, V.V. β-CIT SPECT demonstrates decreased brain dopamine and serotonin transporter levels in untreated parkinsonian patients. Mov. Disord. 2001, 16, 124–130.
- Kim, S.E.; Choi, J.Y.; Choe, Y.S.; Choi, Y.; Lee, W.Y. Serotonin transporters in the midbrain of Parkinson’s disease patients: A study with 123I-β-CIT SPECT. J. Nucl. Med. 2003, 44, 870–876.
- Caretti, V.; Stoffers, D.; Winogrodzka, A.; Isaias, I.U.; Costantino, G.; Pezzoli, G.; Ferrarese, C.; Antonini, A.; Wolters, E.C.; Booij, J. Loss of thalamic serotonin transporters in early drug-naïve Parkinson’s disease patients is associated with tremor: An β-CIT SPECT study. J. Neural Transm. 2008, 115, 721–729.
- Roselli, F.; Pisciotta, N.M.; Pennelli, M.; Aniello, M.S.; Gigante, A.; De Caro, M.F.; Ferrannini, E.; Tartaglione, B.; Niccoli-Asabella, A.; Defazio, G.; et al. Midbrain SERT in degenerative parkinsonisms: A 123I-FP-CIT SPECT study. Mov. Disord. 2010, 25, 1853–1859.
- Qamhawi, Z.; Towey, D.; Shah, B.; Pagano, G.; Seibyl, J.; Marek, K.; Borghammer, P.; Brooks, D.J.; Pavese, N. Clinical correlates of raphe serotonergic dysfunction in early Parkinson’s disease. Brain 2015, 138, 2964–2973.
- Kerenyi, L.; Ricaurte, G.A.; Schretlen, D.J.; McCann, U.; Varga, J.; Mathews, W.B.; Ravert, H.T.; Dannals, R.F.; Hilton, J.; Wong, D.F.; et al. Positron emission tomography of striatal serotonin transporters in Parkinson disease. Arch. Neurol. 2003, 60, 1223–1229.
- Guttman, M.; Boileau, I.; Warsh, J.; Saint-Cyr, J.A.; Ginovart, N.; McCluskey, T.; Houle, S.; Wilson, A.; Mundo, E.; Rusjan, P.; et al. Brain serotonin transporter binding in non-depressed patients with Parkinson’s disease. Eur. J. Neurol. 2007, 14, 523–528.
- Albin, R.L.; Koeppe, R.A.; Bohnen, N.I.; Wernette, K.; Kilbourn, M.A.; Frey, K.A. Spared caudal brainstem SERT binding in early Parkinson’s disease. J. Cereb. Blood Flow Metab. 2008, 28, 441–444.
- Halliday, G.M.; Blumbergs, P.C.; Cotton, R.G.; Blessing, W.W.; Geffen, L.B. Loss of brainstem serotonin- and substance P-containing neurons in Parkinson’s disease. Brain Res. 1990, 510, 104–107.
- Halliday, G.M.; Li, Y.W.; Blumbergs, P.C.; Joh, T.H.; Cotton, R.G.; Howe, P.R.; Blessing, W.W.; Geffen, L.B. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson’s disease. Ann. Neurol. 1990, 27, 373–385.
- Kish, S.J.; Tong, J.; Hornykiewicz, O.; Rajput, A.; Chang, L.J.; Guttman, M.; Furukawa, Y. Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain 2008, 131, 120–131.
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Oertel, W.H.; Björklund, A.; Lindvall, O.; Piccini, P. Serotonin neuron loss and nonmotor symptoms continue in Parkinson’s patients treated with dopamine grafts. Sci. Transl. Med. 2012, 4, 128ra41.
- Del Tredici, K.; Rüb, U.; De Vos, R.A.; Bohl, J.R.; Braak, H. Where does Parkinson’s disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. 2002, 61, 413–426.
- Parkkinen, L.; Pirttila, T.; Alafuzoff, I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008, 115, 399–407.
- Akhtar, R.S.; Stern, M.B. New concepts in the early and preclinical detection of Parkinson’s disease: Therapeutic implications. Expert. Rev. Neurother. 2012, 12, 1429–1438.
- Darweesh, S.K.; Verlinden, V.J.; Stricker, B.H.; Hofman, A.; Koudstaal, P.J.; Ikram, M.A. Trajectories of prediagnostic functioning in Parkinson’s disease. Brain 2017, 140, 429–441.
- Faivre, F.; Joshi, A.; Bezard, E.; Barrot, M. The hidden side of Parkinson’s disease: Studying pain, anxiety and depression in animal models. Neurosci. Biobehav. Rev. 2019, 96, 335–352.
- Bové, J.; Perier, C. Neurotoxin-based models of Parkinson’s disease. Neuroscience 2012, 211, 51–76.
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529.
- Volta, M.; Melrose, H. LRRK2 mouse models: Dissecting the behavior, striatal neurochemistry and neurophysiology of PD pathogenesis. Biochem. Soc. Trans. 2017, 45, 113–122.
- Bastías-Candia, S.; Zolezzi, J.M.; Inestrosa, N.C. Revisiting the Paraquat-Induced Sporadic Parkinson’s Disease-Like Model. Mol. Neurobiol. 2019, 56, 1044–1055.
- Creed, R.B.; Goldberg, M.S. New Developments in Genetic rat models of Parkinson’s Disease. Mov. Disord. 2018, 33, 717–729.
- Francardo, V. Modeling Parkinson’s disease and treatment complications in rodents: Potentials and pitfalls of the current options. Behav. Brain Res. 2018, 352, 142–150.
- Deusser, J.; Schmidt, S.; Ettle, B.; Plötz, S.; Huber, S.; Müller, C.P.; Masliah, E.; Winkler, J.; Kohl, Z. Serotonergic dysfunction in the A53T alpha-synuclein mouse model of Parkinson’s disease. J. Neurochem. 2015, 135, 589–597.
- Björklund, A.; Nilsson, F.; Mattsson, B.; Hoban, D.B.; Parmar, M.A. Combined α-Synuclein/Fibril (SynFib) Model of Parkinson-Like Synucleinopathy Targeting the Nigrostriatal Dopamine System. J. Parkinsons Dis. 2022. preprint.
- Wan, O.W.; Shin, E.; Mattsson, B.; Caudal, D.; Svenningsson, P.; Björklund, A. α-Synuclein induced toxicity in brain stem serotonin neurons mediated by an AAV vector driven by the tryptophan hydroxylase promoter. Sci. Rep. 2016, 6, 26285.
- Jinsmaa, Y.; Cooney, A.; Sullivan, P.; Sharabi, Y.; Goldstein, D.S. The serotonin aldehyde, 5-HIAL, oligomerizes alpha-synuclein. Neurosci. Lett. 2015, 590, 134–137.
- Falsone, S.F.; Leitinger, G.; Karner, A.; Kungl, A.J.; Kosol, S.; Cappai, R.; Zangger, K. The neurotransmitter serotonin interrupts α-synuclein amyloid maturation. Biochim. Biophys. Acta 2011, 1814, 553–561.
- Tin, G.; Mohamed, T.; Shakeri, A.; Pham, A.T.; Rao, P.P.N. Interactions of Selective Serotonin Reuptake Inhibitors with β-Amyloid. ACS Chem. Neurosci. 2019, 10, 226–234.
- Cirrito, J.R.; Wallace, C.E.; Yan, P.; Davis, T.A.; Gardiner, W.D.; Doherty, B.M.; King, D.; Yuede, C.M.; Lee, J.M.; Sheline, Y.I. Effect of escitalopram on Aβ levels and plaque load in an Alzheimer mouse model. Neurology 2020, 95, e2666–e2674.