Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Chondrosarcoma is a malignant cartilaginous tumor that is particularly chemoresistant and radioresistant to X-rays. The first line of treatment is surgery, though this is almost impossible in some specific locations. Such resistances can be explained by the particular composition of the tumor, which develops within a dense cartilaginous matrix, producing a resistant area where the oxygen tension is very low.
- chondrosarcoma
- radiation resistance
- hadrontherapy
1. Introduction
1.1. Overview
Chondrosarcoma is a malignant cartilage tumor of the bone that accounts for about 20–30% of all primary bone sarcomas. This tumor generally affects adults between the ages of 30 and 60 and develops in the limb cavity or on the surface of the bone [1][2][3]. Chondrosarcomas are uncommon in children and adolescents and represent less than 5% of total chondrosarcomas [4]. Chondrosarcoma can develop de novo or from benign cartilage tumors of the bone such as osteochondromas and enchondromas [1]. Anatomically, it is located in the pelvic area, most frequently the ilium, followed by the proximal femur, proximal humerus, distal femur, and ribs. The symptoms can last a long period of time, from months to years, and include pain, pathologic fracture, and common lung metastasis [5]. The number of reported cases in the skull is low; tumors in the nasal cavity are especially rare. This type of chondrosarcoma is more common in children than in adults, with the youngest reported patient aged two years [6]. Due to the location, complete resection is difficult to obtain, and a recurrence of the tumor is quite common.
According to the 2020 World Health Organization, chondrosarcomas are grouped in the malignant category and are classified into three main grades I–III, based on cellularity, tumor matrix characteristics, nuclear features, and mitotic rate [7][8]. Grade I chondrosarcomas are comprised of few cells with no variation in shape and size, and a low metastatic potential associated with flat bone. Both grade II and III chondrosarcomas present hyper-cellularity, variation in cell morphology, and invasion of surrounding tissue. Grade II chondrosarcoma presents a myxoid component while grade III chondrosarcoma is characterized by intense mitotic activity [7][8][9][10].
Chondrosarcomas can also be divided into three subtypes: central, peripheral, and periosteum. The most common type, which happens in about 70% of cases in the proximal femur or proximal humerus, is central chondrosarcoma. The long bones, pelvis, and scapular belt are usually affected by peripheral chondrosarcoma and can develop from pre-existing osteochondromas while periosteal chondrosarcoma can develop on the surface of the bone [1][11][12]. Chondrosarcoma can be classified into lesser-known subtypes such as dedifferentiated, mesenchymal, clear cell, and extra-skeletal myxoid chondrosarcoma [13]. Dedifferentiated chondrosarcomas develop from lower-grade common chondrosarcoma, while mesenchymal chondrosarcoma is very malignant and has a biomorphic histological model with small, round cell islands that interpenetrate with cartilage and dedifferentiate spindle cells [13][14]. Clear-cell chondrosarcoma is less aggressive and is composed of cells containing a large amount of glycogen in the cytoplasm [13][15]. Extra-skeletal myxoid chondrosarcoma is a soft extraskeletal tissue with uncertain differentiation, a predilection for extremities, and a low growth rate [13][16].
1.2. Genetic Characteristics of Chondrosarcoma
Chondrosarcoma relapses and metastases often, thus it is important to identify biomarkers that determine the best clinical approach. Diagnosing chondrosarcomas can be challenging due to its rather common symptoms, low incidence, and grading system [17]. As such, making a correct diagnosis is a key factor for the overall treatment of the tumor. Recent development in imaging methods, endoscopic techniques, gene analysis, biomarker detection, and immunological, and surgical approaches have reduced diagnostic delays [18]. The most commonly used method of diagnostic is radiography, though other imaging methods such as tomography scan, magnetic resonance imaging, bone scintigraphy, and positron emission tomography have been used as adjuvants for patient evaluation [19].
The most frequent mutations found in chondrosarcomas are on isocitrate dehydrogenase (IDH) genes, on arginines R132 for IDH1 and R140/R172 for IDH2 [20]. These genes codes for Krebs cycle enzymes are responsible for the conversion of isocitrate into α-ketoglutarate (α-KG). These mutations induce a gain in function of these enzymes, which can then transform α-KG into an oncometabolite, D-2-hydroxyglutarate (D-2-HG) [21]. Decreased α-KG and increased D-2-HG are associated with epigenetic modifications, such as decreased DNA methylation and hypermethylation of histones associated with differentiation [22]. These changes can also impact the microenvironment since they can impact collagen maturation [23] and block the activity of prolyl hydroxylases [24] thus inducing the stabilization of HIF-1α [25] and HIF-2α [26], responsible for adaptation to hypoxia. The expression of these factors, independently of O2 concentrations, will be responsible for tumor progression and radioresistance. In addition, chondrosarcoma cells are often EXT1/2 mutated and CDKN2A/B deletions are also frequent, as well as COL2A1 mutations [27]. In osteosarcoma, another rare malignant bone tumor, the expression of several repetitive elements was observed differentially expressed with normal bone. HERVs’ (human endogenous retroviruses) integrated sequences and satellite elements were the most significantly differential expressed elements between osteosarcoma and normal tissues and could help to understand the genesis mechanism of such sarcoma [28]. In addition, a transcriptomic analysis of osteosarcoma bone samples revealed that BTNL9, MMP14, ABCA10, ACACB, COL11A1, and PKM2 were expressed differentially with the highest significance between tumor and normal bone, and reflected the changes in the regulation of the degradation of collagen and extracellular matrix [29].
1.3. Chondrosarcoma Standard Treatment
According to ESMO guidelines [30], standard treatment depends on the localization, the histological subtype, and the level of differentiation. Grade I low-grade peripheral chondrosarcoma (arising from osteochondroma) or grade I central chondrosarcoma of the long bones, can be managed with minimally-invasive surgery (curetage for example) without adjuvant treatment. On the opposite higher-grade chondrosarcoma, such as clear cell chondrosarcoma, mesenchymal chondrosarcoma, and dedifferentiated or axial chondrosarcoma (pelvic, spine, or skull base) should be extensively resected with wide margins and postoperative irradiation is often proposed in case R1 or R2 resections, or for the management of local recurrences. For R1 and R2 low-grade skull-base chondrosarcomas, the best timing for irradiation remains to be discussed. Immediate or delayed irradiation could be proposed according to each center’s policies. Recent evidence suggests that mesenchymal chondrosarcoma may be chemotherapy sensitive, and may be considered for adjuvant or neoadjuvant therapy [30].
It is known that chondrosarcoma is a chemo- and radioresistant tumor [31]. Slow proliferation, overexpression of the protein involved in drug resistance MDR1, poor vascularization, and dense extracellular matrix may be responsible for chemo- and radioresistance of chondrosarcoma [32]
Although many studies have obtained good results, monotherapy of heterogeneous chondrosarcoma is a concern due to the tumor’s ability to adapt. Moreover, the failure of a monotherapy should not exclude its potential for being an adjuvant therapy in combined treatment. The use of novel nonconventional therapies can improve the outcome, though the adverse effects should be taken into consideration and further clinical investigations are required to assess the safety and efficacy in a large group of patients.
1.3.1. Chondrosarcoma Chemotherapy
Chemotherapy is rarely effective and the studies on patients are limited due to the rarity of these diseases [32]. As such, current treatments have a base in ostosarcoma treatment [33]. As such, palliative treatments with cisplatin, doxorubicin, or ifosfamide are also used in clinical treatments [34] even though chondrosarcoma has presented resistance to doxorubicin in vitro [35].
Nonconventional treatments for chondrosarcoma include molecularly-targeted therapies, epigenetic approaches, immunotherapy, and herbal therapies [33][36]. Some of the targets for chondrosarcoma therapies involve mutations of isocitrate dehydrogenases IDH1 and IDH2 [37][38][39][40], angiogenesis [41][42], cyclin-dependent kinases (CDK) [43], tyrosine kinase inhibitor [44], mechanisms involved in the signaling pathway of Rapamycin (mTOR) [45][46], agents of hypomethylation, and histone deacetylase (HDAC) [47][48][49]. Clinical studies involving immune checkpoint inhibition are in the early stages and show promising results despite patients’ mixed responses to some inhibitors [50][51][52]. A large and complete overview of new targeted therapies in chondrosarcoma can be found in two recent review studies by Boehme et al. [32] and Tlemsani et al. [27].
1.3.2. Chondrosarcoma Radiotherapy
Conventional radiation therapy is used for patients with incomplete resections, inoperable tumors, or metastases [53][54][55].
Photon radiotherapy has low accuracy, with undesirable toxicity in the surrounding normal tissue, and is not suitable for targeting tumors larger than 2.5 cm [56]. Over the past decade, radiation therapy has developed better control over localization and dosage with a limited effect on surrounding healthy tissue [57]. There are several types of radiological techniques used to treat inoperable tumors such as external radiation/source therapy, modified fraction radiotherapy, internal radiation therapy, and particle therapy. Therefore, each type of radiation therapy machine has distinct physical characteristics, which may influence clinical outcomes [58].
Clinical studies have shown poor results from low-dose radiation therapy. Indeed, delivering a dose higher than 70 Gy is mandatory to obtain local control. This dose is difficult to reach with conventional radiotherapy due to surrounding neural structures for which the tolerance dose is well below 70 Gy (from 54 to 60 Gy for the optic tract or brainstem, and 50 Gy for the spinal cord, for example). Modern techniques such as intensive modulated radiotherapy (IMRT), stereotactic radiosurgery, and hadron therapy can overcome these limits [59]. Reports show long-term promising results in patients with spine tumors when using a combination of IMPT and IMRT [60] or for patients with intracranial chondrosarcoma undergoing stereotactic radiosurgery [61]. Survival rates depend on the dose, age, tumor size, and quality of the surgical treatment. An improvement in radiation delivery remains necessary to increase the therapeutic ratio [55].
2. Radiation Resistance of Chondrosarcoma: Microenvironment, Molecular and Cellular Consequences
As previously mentioned, chondrosarcoma is particularly resistant to conventional radiotherapy, especially due to its very dense cartilaginous extracellular matrix and the presence of some cells in the tumor tissue that may proliferate slowly, whereas radiotherapy (RT) is more effective on rapidly dividing cells [62].
2.1. Hypoxia-Related Radiation Resistance
The characteristics mentioned above, and the low proportion of blood vessels in these tumors, also lead to a hypoxic microenvironment in the tumor. It is widely described that the decrease in oxygen content causes a reduction in the effectiveness of X-ray RT. Indeed, the lethal effects of X-rays are triggered by indirect DNA damage, which is mainly caused by the formation of reactive oxygen species (ROS) due to the radiolysis of water and dissolved oxygen [63]. Thus, hypoxia leads directly to a decrease in the efficiency of RT via an absence of O2 concentrations (Figure 1).
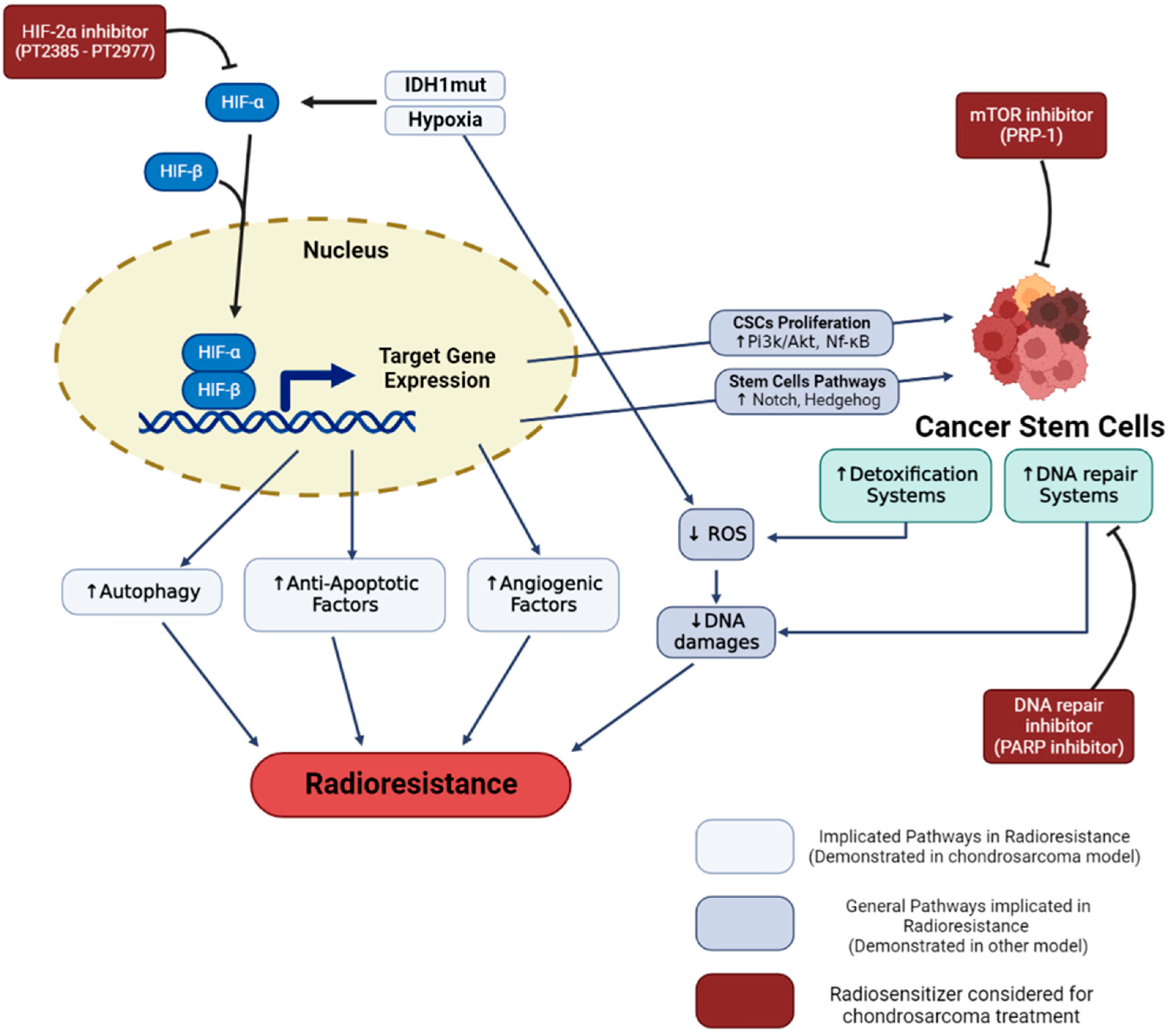
Figure 1. Hypoxia and cancer stem cells radioresistance in chondrosarcoma. Hypoxia and IDH-1 mutation lead to HIF-1α and HIF-2α stabilization. These regulated pathways enhance cell radioresistance and increase the proportion of cancer stem cells. Specific inhibitors (in red), such as HIF-2, PARP, and mTOR inhibitors demonstrated a capacity to reverse cell resistance and are considered in order to radiosensitize chondrosarcomas.
Hypoxia allows an adaptation of the cancer cells which can give rise to radioresistance. Different signaling pathways have been described, such as autophagy increase [64], stabilization and signaling of the HIF-1 factor, increased secretion of exosomes, or reprogramming of energy metabolism [65].
Concerning chondrosarcoma, HIF-1α stabilization is observed when cells are cultured in hypoxia [66]. The expression of HIF-1α notably allows the activation of angiogenesis pathways, via the expression of vascular endothelial growth factor (VEGF) [66][67]. In the clinic, the high expression of the hypoxia factor HIF1-α is notably associated with poor prognosis and metastatic tumors with poor patient survival. This suggests that activation of the transcription factor HIF-1α may play a role in tumor progression [68][69]. More recent studies have shown that HIF-2α also plays an important role in the progression of chondrosarcoma cells by promoting tumor-initiating and invasive properties [26]. In addition, mutations found in chondrosarcoma such as mutations located in IDH1/2 (>50% of chondrosarcomas) have been described to induce constitutive activation of HIF-1α and Hif-2α factors [25][26]. Although hypoxia is known to induce radioresistance in other cancer cell models, very few publications have been able to make this link directly in chondrosarcoma. For example, one study showed that overexpression of HIF-1α was associated with an overexpression of BCL-xl (a bcl2 family factor), notably conducting to antiapoptotic properties that can generate chemo- and radioresistance [70]. In closely related models, such as osteosarcoma, it has been shown that hypoxia can induce increased autophagy in connection with radioresistance [64].
The hypoxic microenvironment can also cause a form of resistance, by favoring the presence of more resistant cellular subtypes, the cancer stem cells (CSCs) [71]. Indeed, it has been described that the factors HIF-1α and HIF-2α allow the proliferation of CSCs via the activation of the PI3K/AKT [72] and NF-κB [73] pathways. These hypoxia factors enable the activation of the notch [74][75], and hedgehog [76] pathways, responsible for maintaining the stem potential of this cell subtype [77].
2.2. Radioresistance Links with CSCs
Cancer stem cells (CSCs) are a subpopulation of cancer cells within the tumor that have been associated with treatment resistance, tumor relapse, and metastasis in several cancers including chondrosarcoma (CS). CSC (or tumor-initiating cells) are seen as drivers of tumor establishment and growth, often correlated to aggressive, heterogeneous, and therapy-resistant tumors [78]. The concept of CSC is also related to specific cellular biomarkers. Indeed, several markers expressed in CSCs can also be found in adult tissue-resident stem cell populations [79]. In order to describe this particular type of cell, the term of CSCs was kept, knowing that it could be the subject of controversial theories [80][81]. CSCs are defined as dedifferentiated cells that have unlimited proliferation and self-renewal abilities and can reinitiate and reconstitute tumor heterogeneity [82]. CSCs have inherited normal stem cells properties, including a hypoxic niche that protects them from treatments and promotes the quiescence state, telomerase activity, an increase in the activity of membrane transporters and detoxification enzyme, activation of antiapoptotic pathways, an enhanced DNA repair capacity, and catabolism of ROS (Figure 1). These properties will block or reduce the cellular effects induced by actual antitumor treatments [2][3]. These persisting cells, even few in number, can proliferate and reconstitute the tumor with all its phenotypic diversity. Resistance to therapeutic treatment, such as chemotherapy and radiotherapy, could be associated with the fact that current therapies do not target CSCs [83].
Several CSC identification and isolation methods have been described [84]. In vitro assays are commonly used to isolate CSCs such as the sphere formation assay called tumorspheres, under nonadherent, serum-free conditions and enriched with growth factors. In vivo, the isolated CSCs are transplanted in immunocompromised mice to assess the tumorigenic capacity of the CSCs. As CSCs have the same properties as normal stem cells, they can be identified within the tumors with normal stem cell markers such as SOX2, OCT3/4, Nanog, or Nestin. They are also identified in multiple solid tumors by means of the CSC’s specific cell surface markers and side population phenotype [85].
In CS, CSCs are commonly characterized by molecular markers such as aldehyde dehydrogenase (ALDH) and prominin-1 (CD133) [86]. ALDH is an enzyme that oxidizes aldehydes to carboxylic acids and allows cells to resist oxidative stress. Every cell expresses ALDH, however, cells with high ALDH activity have demonstrated enhanced tumorigenicity in several cancer cell types, characteristic of CSCs [87]. CD133 is a transmembrane glycoprotein, and its exact function remains to be elucidated, though it seems to be involved in membrane organization, cell differentiation, proliferation, and signal transduction. In addition, it might also have a role in apoptosis inhibition, and the upregulation of FLICE-like inhibitory protein (FLIP), leading to chemoresistance [88]. This cell-surface protein is known as an important driver of tumor progression and as a CSC marker [89].
Several studies identify CS CSC as ALDH+ and CD133+ cells and it was considered that the combination of ALDH+ CD133+ was the best marker to identify the tumor population enriched with the CSC phenotype [87][90]. Tirino et al. provided evidence of the presence of CSC in human primary bone sarcoma and demonstrated the possibility to use the CD133 marker for their identification [85]. They showed that CD133 was expressed in 21 fresh biopsies from bone and soft tissue sarcomas. After sorting cells, the CD133+ cells were able to reconstitute the original cell population, demonstrating the capacity of CSCs to dedifferentiate and rebuild tumor heterogeneity. Furthermore, they showed that the CD133+ cells were able to form tumorspheres. These spheres were positive for CD133 and the transcription factors OCT3/4, Nanog, Sox-2, and Nestin, which are involved in self-renewal and in the preservation of pluri-multipotency of normal stem cells. They demonstrated the ability of this cell population to differentiate into adipocytes and osteoblasts, supporting the fact that they originate from the mesenchymal stem cells of bone sarcomas. In addition, they showed in vivo that the cell population was capable of generating tumors in mice. These different evidences proved that the CD133+ sorted cells were CSCs, and, thus, CD133 is a useful marker for the identification of the CSC population. Greco et al. showed a significant correlation between ALDH activity and metastatic potential in ten patients with bone sarcoma, including CS. Moreover, they proved that bone sarcoma cells were sensitive to ALDH inhibition with disulfiram, involving a potential use of ALDH inhibition as a therapeutic strategy for radio and chemoresistant CS, although more investigations are required [87].
One of the main features of the CSC subpopulation is the overexpression of transcription factors such as SOX2 or OCT4, involved in the maintenance of stem cell phenotype in normal stem cells. In sarcomas, including CS, SOX2 has been found overexpressed in CSCs [91]. The same observations were made in relation to OCT4 in osteosarcomas and Ewing sarcoma, close models of CS. Menendez et al. introduced a system to monitor the transcriptional activity of SOX2 and OCT4 (SORE6) in CS patient-derive cell lines, in vitro and immunodeficient mice [91]. The system allows for isolating SOX2/OCT4 positive cells and thus analyzing the tumor-promoting CSC in sarcoma. They detected 20% of the SORE6+ cells, and this percentage was found increased to 40% in immunodeficient mice, which could be due to the elevation of the CSCs during tumor progression and adaptation to new microenvironments. They also showed that CSC-related genes, including SOX2, were overexpressed in the tumorsphere and enhanced during sarcoma progression. These results proved that SOX2 can be used as a CSC marker in sarcomas. Moreover, the system SORE6 is a good tool to evaluate the activity of antitumor drugs on CSC [91].
Another way to identify the CSC population is the so-called side population. CSC can evade treatment potentially through the increase in ATP-binding cassette (ABC) multidrug efflux transporters such as MDR1/ABCB1, BRCP1/ABCG2, and ABCB5 expression. The ability of cells to exclude DNA-binding dyes is measured. A side population appears as the cells expressing high ABC transporters exclude the dyes [84]. This was studied in osteosarcoma, however, it would be interesting to test this assay in the CS model.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15071962
References
- Nazeri, E.; Gouran Savadkoohi, M.; Majidzadeh-A, K.; Esmaeili, R. Chondrosarcoma: An Overview of Clinical Behavior, Molecular Mechanisms Mediated Drug Resistance and Potential Therapeutic Targets. Crit. Rev. Oncol./Hematol. 2018, 131, 102–109.
- Dai, X.; Ma, W.; He, X.; Jha, R.K. Review of Therapeutic Strategies for Osteosarcoma, Chondrosarcoma, and Ewing’s Sarcoma. Med. Sci. Monit. 2011, 17, RA177-190.
- David, E.; Blanchard, F.; Heymann, M.F.; De Pinieux, G.; Gouin, F.; Rédini, F.; Heymann, D. The Bone Niche of Chondrosarcoma: A Sanctuary for Drug Resistance, Tumour Growth and Also a Source of New Therapeutic Targets. Sarcoma 2011, 2011, 932451.
- Puri, A. Chondrosarcomas in Children and Adolescents. EFORT Open Rev. 2020, 5, 90–95.
- Ottesen, T.D.; Shultz, B.N.; Munger, A.M.; Amick, M.; Toombs, C.S.; Friedaender, G.E.; Grauer, J.N. Chondrosarcoma Patient Characteristics, Management, and Outcomes Based on over 5000 Cases from the National Cancer Database (NCDB). PLoS ONE 2022, 17, e0268215.
- Chou, P.; Mehta, S.; Gonzalez-Crussi, F. Chondrosarcoma of the head in children. Pediatr. Pathol. 1990, 10, 945–958.
- Tosun, I.; Naderi, S. Approach to Primary Vertebral Tumors in the Light of the 2020 Updated World Health Organization Classification of Bone Tumors. Turk. Neurosurg. 2021.
- Zoccali, C.; Baldi, J.; Attala, D.; Rossi, B.; Anelli, V.; Annovazzi, A.; Ferraresi, V. Intralesional vs. Extralesional Procedures for Low-Grade Central Chondrosarcoma: A Systematic Review of the Literature. Arch. Orthop. Trauma Surg. 2018, 138, 929–937.
- Fletcher, C.D.; Unni, K.K.; Mertens, F. Pathology and Genetics of Tumours of Soft Tissue and Bone; International Agency for Research on Cancer: Lyon, France, 2002; Volume 177, ISBN 92-832-2413-2.
- Girard, N.; Lhuissier, E.; Aury-Landas, J.; Cauvard, O.; Lente, M.; Boittin, M.; Baugé, C.; Boumédiene, K. Heterogeneity of Chondrosarcomas Response to Irradiations with X-rays and Carbon Ions: A Comparative Study on Five Cell Lines. J. Bone Oncol. 2020, 22, 100283.
- Righi, A.; Pacheco, M.; Cocchi, S.; Asioli, S.; Gambarotti, M.; Donati, D.M.; Evangelista, A.; Gnoli, M.; Locatelli, M.; Mordenti, M.; et al. Secondary Peripheral Chondrosarcoma Arising in Solitary Osteochondroma: Variables Influencing Prognosis and Survival. Orphanet. J. Rare Dis. 2022, 17, 74.
- Chaabane, S.; Bouaziz, M.C.; Drissi, C.; Abid, L.; Ladeb, M.F. Periosteal Chondrosarcoma. Am. J. Roentgenol. 2009, 192, W1–W6.
- Chow, W.A. Chondrosarcoma: Biology, Genetics, and Epigenetics . F1000Research 2018, 7, 1826.
- Strach, M.C.; Grimison, P.S.; Hong, A.; Boyle, R.; Stalley, P.; Karim, R.; Connolly, E.A.; Bae, S.; Desai, J.; Crowe, P.; et al. Mesenchymal Chondrosarcoma: An Australian Multi-Centre Cohort Study. Cancer Med. 2022, 12, 368–378.
- Klein, A.; Tauscher, F.; Birkenmaier, C.; Baur-Melnyk, A.; Knösel, T.; Jansson, V.; Dürr, H.R. Clear Cell Chondrosarcoma Is an Underestimated Tumor: Report of 7 Cases and Meta-Analysis of the Literature. J. Bone Oncol. 2019, 19, 100267.
- Stacchiotti, S.; Baldi, G.G.; Morosi, C.; Gronchi, A.; Maestro, R. Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management. Cancers 2020, 12, 2703.
- Cozma, G.V.; Sima, L.V.; Cloşca, R.M.; Baderca, F.; Horhat, I.D.; Balica, N.C.; Tischer, A.A.; Moţ, I.C.; Maliţa, D.C.; Marin, A.; et al. Conventional Grade 1 Chondrosarcoma: A Challenging Diagnosis with Important Implications on Therapy and Prognosis. Rom. J. Morphol. Embryol. 2021, 62, 605–613.
- Hosseini, A.; Mirzaei, A.; Salimi, V.; Jamshidi, K.; Babaheidarian, P.; Fallah, S.; Rampisheh, Z.; Khademian, N.; Abdolvahabi, Z.; Bahrabadi, M.; et al. The Local and Circulating SOX9 as a Potential Biomarker for the Diagnosis of Primary Bone Cancer. J. Bone Oncol. 2020, 23, 100300.
- Zhang, Q.; Xi, Y.; Li, D.; Yuan, Z.; Dong, J. The Utility of 18F-FDG PET and PET/CT in the Diagnosis and Staging of Chondrosarcoma: A Meta-Analysis. J. Orthop. Surg. Res. 2020, 15, 229.
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 Mutations Are Frequent Events in Central Chondrosarcoma and Central and Periosteal Chondromas but Not in Other Mesenchymal Tumours. J. Pathol. 2011, 224, 334–343.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744.
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature 2012, 483, 474–478.
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-Hydroxyglutarate Produced by Mutant IDH1 Perturbs Collagen Maturation and Basement Membrane Function. Genes Dev. 2012, 26, 2038–2049.
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1alpha. Science 2009, 324, 261–265.
- Hu, X.; Li, L.; Eid, J.E.; Liu, C.; Yu, J.; Yue, J.; Trent, J.C. IDH1 Mutation Induces HIF-1α and Confers Angiogenic Properties in Chondrosarcoma JJ012 Cells. Dis. Mrk. 2022, 2022, 7729968.
- Kim, H.; Cho, Y.; Kim, H.-S.; Kang, D.; Cheon, D.; Kim, Y.-J.; Chang, M.J.; Lee, K.M.; Chang, C.B.; Kang, S.-B.; et al. A System-Level Approach Identifies HIF-2α as a Critical Regulator of Chondrosarcoma Progression. Nat. Commun. 2020, 11, 5023.
- Tlemsani, C.; Larousserie, F.; De Percin, S.; Audard, V.; Hadjadj, D.; Chen, J.; Biau, D.; Anract, P.; Terris, B.; Goldwasser, F.; et al. Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options. Int. J. Mol. Sci. 2023, 24, 1361.
- Ho, X.D.; Nguyen, H.G.; Trinh, L.H.; Reimann, E.; Prans, E.; Kõks, G.; Maasalu, K.; Le, V.Q.; Nguyen, V.H.; Le, N.T.N.; et al. Analysis of the Expression of Repetitive DNA Elements in Osteosarcoma. Front. Genet. 2017, 8, 193.
- Ho, X.D.; Phung, P.; Le, V.Q.; Nguyen, V.H.; Reimann, E.; Prans, E.; Kõks, G.; Maasalu, K.; Le, N.T.; Trinh, L.H.; et al. Whole Transcriptome Analysis Identifies Differentially Regulated Networks between Osteosarcoma and Normal Bone Samples. Exp. Biol. Med. 2017, 242, 1802–1811.
- Hogendoorn, P.C.W.; Athanasou, N.; Bielack, S.; Alava, E.D.; Tos, A.P.D.; Ferrari, S.; Gelderblom, H.; Grimer, R.; Hall, K.S.; Hassan, B.; et al. Bone Sarcomas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2010, 21, v204–v213.
- Jeong, W.; Kim, H.J. Biomarkers of Chondrosarcoma. J. Clin. Pathol. 2018, 71, 579–583.
- Boehme, K.A.; Schleicher, S.B.; Traub, F.; Rolauffs, B. Chondrosarcoma: A Rare Misfortune in Aging Human Cartilage? The Role of Stem and Progenitor Cells in Proliferation, Malignant Degeneration and Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 311.
- Monga, V.; Mani, H.; Hirbe, A.; Milhem, M. Non-Conventional Treatments for Conventional Chondrosarcoma. Cancers 2020, 12, 1962.
- Whelan, J.S.; Davis, L.E. Osteosarcoma, Chondrosarcoma, and Chordoma. J. Clin. Oncol. 2018, 36, 188–193.
- Gallego, B.; Murillo, D.; Rey, V.; Huergo, C.; Estupiñán, Ó.; Rodríguez, A.; Tornín, J.; Rodríguez, R. Addressing Doxorubicin Resistance in Bone Sarcomas Using Novel Drug-Resistant Models. Int. J. Mol. Sci. 2022, 23, 6425.
- MacDonald, I.J.; Lin, C.Y.; Kuo, S.J.; Su, C.M.; Tang, C.H. An Update on Current and Future Treatment Options for Chondrosarcoma. Expert Rev. Anticancer Ther. 2019, 19, 773–786.
- Shah, F.H.; Kim, S.J. Identification of Medicinal Compounds as Potential Inhibitors for Mutated Isocitrate Dehydrogenases against Chondrosarcoma. Saudi J. Biol. Sci. 2022, 29, 161–167.
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Therapeutic Targets and Emerging Treatments in Advanced Chondrosarcoma. Int. J. Mol. Sci. 2022, 23, 1096.
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Ivosidenib, an Oral, Targeted Inhibitor of Mutant IDH1, in Patients with Advanced Solid Tumors. Investig. New Drugs 2020, 38, 433–444.
- Khurshed, M.; Molenaar, R.J.; van Linde, M.E.; Mathôt, R.A.; Struys, E.A.; van Wezel, T.; van Noorden, C.J.F.; Klümpen, H.J.; Bovée, J.V.M.G.; Wilmink, J.W. A Phase Ib Clinical Trial of Metformin and Chloroquine in Patients with Idh1-Mutated Solid Tumors. Cancers 2021, 13, 2474.
- Jones, R.L.; Katz, D.; Loggers, E.T.; Davidson, D.; Rodler, E.T.; Pollack, S.M. Clinical Benefit of Antiangiogenic Therapy in Advanced and Metastatic Chondrosarcoma. Med. Oncol. 2017, 34, 167.
- Chow, W.; Frankel, P.; Ruel, C.; Araujo, D.M.; Milhem, M.; Okuno, S.; Hartner, L.; Undevia, S.; Staddon, A. Results of a Prospective Phase 2 Study of Pazopanib in Patients with Surgically Unresectable or Metastatic Chondrosarcoma. Cancer 2020, 126, 105–111.
- Shen, J.; Yu, C.; Wang, Z.; Mu, H.; Cai, Z. PLCD1-Induced DNA Damage Inhibits the Tumor Growth via Downregulating CDKs in Chondrosarcoma. J. Oncol. 2022, 2022, 4488640.
- O’Sullivan Coyne, G.; Kummar, S.; Hu, J.; Ganjoo, K.; Chow, W.A.; Do, K.T.; Zlott, J.; Bruns, A.; Rubinstein, L.; Foster, J.C.; et al. Clinical Activity of Single-Agent Cabozantinib (XL184), a Multi-Receptor Tyrosine Kinase Inhibitor, in Patients with Refractory Soft-Tissue Sarcomas. Clin. Cancer Res. 2022, 28, 279–288.
- Wang, W.; Yan, T.; Guo, W.; Niu, J.; Zhao, Z.; Sun, K.; Zhang, H.; Yu, Y.; Ren, T. Constitutive GLI1 Expression in Chondrosarcoma Is Regulated by Major Vault Protein via MTOR/S6K1 Signaling Cascade. Cell Death Differ. 2021, 28, 2221–2237.
- van Oosterwijk, J.G.; Anninga, J.K.; Gelderblom, H.; Cleton-Jansen, A.M.; Bovée, J.V.M.G. Update on Targets and Novel Treatment Options for High-Grade Osteosarcoma and Chondrosarcoma. Hematol./Oncol. Clin. North Am. 2013, 27, 1021–1048.
- Hamm, C.A.; Xie, H.; Costa, F.F.; Vanin, E.F.; Seftor, E.A.; Sredni, S.T.; Bischof, J.; Wang, D.; Bonaldo, M.F.; Hendrix, M.J.C.; et al. Global Demethylation of Rat Chondrosarcoma Cells after Treatment with 5-Aza-2′-Deoxycytidine Results in Increased Tumorigenicity. PLoS ONE 2009, 4, e8340.
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767.
- Sheng, W.; Zhang, Z.-C.; Shi, D.-Y.; Wang, B.-C.; Wu, Q.; Shao, Z.-W.; Yang, S.-H.; He, T.-C.; Liu, J.-X. Epigenetic Silencing of SFRP5 Promotes the Metastasis and Invasion of Chondrosarcoma by Expression Inhibition and Wnt Signaling Pathway Activation. Chem.-Biol. Interact. 2018, 296, 1–8.
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in Immune Checkpoint Inhibitors for Bone Sarcoma Therapy. J. Bone Oncol. 2019, 15, 100221.
- Wagner, M.J.; Ricciotti, R.W.; Mantilla, J.; Loggers, E.T.; Pollack, S.M.; Cranmer, L.D. Response to PD1 Inhibition in Conventional Chondrosarcoma. J. Immunother. Cancer 2018, 6, 94.
- Chen, S.; Fritchie, K.; Wei, S.; Ali, N.; Curless, K.; Shen, T.; Brini, A.T.; Latif, F.; Sumathi, V.; Siegal, G.P.; et al. Diagnostic Utility of IDH1/2 Mutations to Distinguish Dedifferentiated Chondrosarcoma from Undifferentiated Pleomorphic Sarcoma of Bone. Hum. Pathol. 2017, 65, 239–246.
- de Jong, Y.; Ingola, M.; Briaire-de Bruijn, I.H.; Kruisselbrink, A.B.; Venneker, S.; Palubeckaite, I.; Heijs, B.P.A.M.; Cleton-Jansen, A.-M.; Haas, R.L.M.; Bovée, J.V.M.G. Radiotherapy Resistance in Chondrosarcoma Cells; a Possible Correlation with Alterations in Cell Cycle Related Genes. Clin. Sarcoma Res. 2019, 9, 9.
- Amichetti, M.; Amelio, D.; Cianchetti, M.; Giacomelli, I.; Scartoni, D. The Treatment of Chordoma and Chondrosarcoma of the Skull Base with Particular Attention to Radiotherapy. Clin. Oncol. 2017, 2, 1195.
- Catanzano, A.A.; Kerr, D.L.; Lazarides, A.L.; Dial, B.L.; Lane, W.O.; Blazer, D.G.; Larrier, N.A.; Kirsch, D.G.; Brigman, B.E.; Eward, W.C. Revisiting the Role of Radiation Therapy in Chondrosarcoma: A National Cancer Database Study. Sarcoma 2019, 2019, 4878512.
- Mehta, S.R.; Suhag, V.; Semwal, M.; Sharma, N. Radiotherapy: Basic Concepts and Recent Advances. Med. J. Armed Forces India 2010, 66, 158–162.
- Lepleux, C.; Marie-Brasset, A.; Temelie, M.; Boulanger, M.; Brotin, É.; Goldring, M.B.; Hirtz, C.; Varès, G.; Nakajima, T.; Saintigny, Y.; et al. Bystander Effectors of Chondrosarcoma Cells Irradiated at Different LET Impair Proliferation of Chondrocytes. J. Cell Commun. Signal. 2019, 13, 343–356.
- Allen, C.; Her, S.; Jaffray, D.A. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2.
- Mercado, C.E.; Holtzman, A.L.; Rotondo, R.; Rutenberg, M.S.; Mendenhall, W.M. Proton Therapy for Skull Base Tumors: A Review of Clinical Outcomes for Chordomas and Chondrosarcomas. Head Neck 2019, 41, 536–541.
- DeLaney, T.F.; Liebsch, N.J.; Pedlow, F.X.; Adams, J.; Weyman, E.A.; Yeap, B.Y.; Depauw, N.; Nielsen, G.P.; Harmon, D.C.; Yoon, S.S.; et al. Long-Term Results of Phase II Study of High Dose Photon/Proton Radiotherapy in the Management of Spine Chordomas, Chondrosarcomas, and Other Sarcomas. J. Surg. Oncol. 2014, 110, 115–122.
- Kano, H.; Niranjan, A.; Dade Lunsford, L. Radiosurgery for Chordoma and Chondrosarcoma. Prog. Neurol. Surg. 2019, 34, 207–214.
- Gelderblom, H.; Hogendoorn, P.C.W.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.M.; Bovée, J.V.M.G. The Clinical Approach towards Chondrosarcoma. The Oncologist 2008, 13, 320–329.
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112.
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-Induced Autophagy as an Additional Mechanism in Human Osteosarcoma Radioresistance. J. Bone Oncol. 2016, 5, 67–73.
- Kabakov, A.E.; Yakimova, A.O. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers 2021, 13, 1102.
- Lin, C.; McGough, R.; Aswad, B.; Block, J.A.; Terek, R. Hypoxia Induces HIF-1Œ± and VEGF Expression in Chondrosarcoma Cells and Chondrocytes. J. Orthop. Res. 2004, 22, 1175–1181.
- McGough, R.L.; Aswad, B.I.; Terek, R.M. Pathologic Neovascularization in Cartilage Tumors. Clin. Orthop. Relat. Res. 2002, 397, 76–82.
- Kubo, T.; Sugita, T.; Shimose, S.; Matsuo, T.; Arihiro, K.; Ochi, M. Expression of Hypoxia-Inducible Factor-1alpha and Its Relationship to Tumour Angiogenesis and Cell Proliferation in Cartilage Tumours. J. Bone Jt. Surg. Br. 2008, 90, 364–370.
- Boeuf, S.; Bovée, J.V.M.G.; Lehner, B.; Hogendoorn, P.C.W.; Richter, W. Correlation of Hypoxic Signalling to Histological Grade and Outcome in Cartilage Tumours. Histopathology 2010, 56, 641–651.
- Chen, C.; Zhou, H.; Wei, F.; Jiang, L.; Liu, X.; Liu, Z.; Ma, Q. Increased Levels of Hypoxia-Inducible Factor-1α Are Associated with Bcl-XL Expression, Tumor Apoptosis, and Clinical Outcome in Chondrosarcoma: Increased Levels Of HIF-1α in Chondrosarcoma. J. Orthop. Res. 2011, 29, 143–151.
- Sun, X.; Lv, X.; Yan, Y.; Zhao, Y.; Ma, R.; He, M.; Wei, M. Hypoxia-Mediated Cancer Stem Cell Resistance and Targeted Therapy. Biomed. Pharm. 2020, 130, 110623.
- Marhold, M.; Tomasich, E.; El-Gazzar, A.; Heller, G.; Spittler, A.; Horvat, R.; Krainer, M.; Horak, P. HIF1α Regulates MTOR Signaling and Viability of Prostate Cancer Stem Cells. Mol. Cancer Res. 2015, 13, 556–564.
- Wu, S.-L.; Li, Y.-J.; Liao, K.; Shi, L.; Zhang, N.; Liu, S.; Hu, Y.-Y.; Li, S.-L.; Wang, Y. 2-Methoxyestradiol Inhibits the Proliferation and Migration and Reduces the Radioresistance of Nasopharyngeal Carcinoma CNE-2 Stem Cells via NF-ΚB/HIF-1 Signaling Pathway Inactivation and EMT Reversal. Oncol. Rep. 2017, 37, 793–802.
- Qian, J.; Rankin, E.B. Hypoxia-Induced Phenotypes That Mediate Tumor Heterogeneity. Adv. Exp. Med. Biol. 2019, 1136, 43–55.
- Yan, Y.; Liu, F.; Han, L.; Zhao, L.; Chen, J.; Olopade, O.I.; He, M.; Wei, M. HIF-2α Promotes Conversion to a Stem Cell Phenotype and Induces Chemoresistance in Breast Cancer Cells by Activating Wnt and Notch Pathways. J. Exp. Clin. Cancer Res. 2018, 37, 256.
- Yan, G.-N.; Yang, L.; Lv, Y.-F.; Shi, Y.; Shen, L.-L.; Yao, X.-H.; Guo, Q.-N.; Zhang, P.; Cui, Y.-H.; Zhang, X.; et al. Endothelial Cells Promote Stem-like Phenotype of Glioma Cells through Activating the Hedgehog Pathway. J. Pathol. 2014, 234, 11–22.
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting Cancer Stem Cells by Inhibiting Wnt, Notch, and Hedgehog Pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106.
- Kuşoğlu, A.; Biray Avcı, Ç. Cancer Stem Cells: A Brief Review of the Current Status. Gene 2019, 681, 80–85.
- Kim, W.-T.; Ryu, C.J. Cancer Stem Cell Surface Markers on Normal Stem Cells. BMB Rep. 2017, 50, 285–298.
- Vermeulen, L.; Sprick, M.R.; Kemper, K.; Stassi, G.; Medema, J.P. Cancer Stem Cells—Old Concepts, New Insights. Cell Death Differ. 2008, 15, 947–958.
- Tu, S.-M.; Estecio, M.R.; Lin, S.-H.; Zacharias, N.M. Stem Cell Theory of Cancer: Rude Awakening or Bad Dream from Cancer Dormancy? Cancers 2022, 14, 655.
- Vares, G.; Ahire, V.; Sunada, S.; Ho Kim, E.; Sai, S.; Chevalier, F.; Romeo, P.-H.; Yamamoto, T.; Nakajima, T.; Saintigny, Y. A Multimodal Treatment of Carbon Ions Irradiation, MiRNA-34 and MTOR Inhibitor Specifically Control High-Grade Chondrosarcoma Cancer Stem Cells. Radiother. Oncol. 2020, 150, 253–261.
- Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F.; Zhu, C. Therapeutic Approaches Targeting Cancer Stem Cells. J. Cancer Res. Ther. 2018, 14, 1469–1475.
- Brown, H.K.; Tellez-Gabriel, M.; Heymann, D. Cancer Stem Cells in Osteosarcoma. Cancer Lett. 2017, 386, 189–195.
- Tirino, V.; Desiderio, V.; Paino, F.; De Rosa, A.; Papaccio, F.; Fazioli, F.; Pirozzi, G.; Papaccio, G. Human Primary Bone Sarcomas Contain CD133+ Cancer Stem Cells Displaying High Tumorigenicity in Vivo. FASEB J. 2011, 25, 2022–2030.
- Rey, V.; Menendez, S.T.; Estupiñan, O.; Rodriguez, A.; Santos, L.; Tornin, J.; Martinez-Cruzado, L.; Castillo, D.; Ordoñez, G.R.; Costilla, S.; et al. New Chondrosarcoma Cell Lines with Preserved Stem Cell Properties to Study the Genomic Drift During In Vitro/In Vivo Growth. JCM 2019, 8, 455.
- Greco, N.; Schott, T.; Mu, X.; Rothenberg, A.; Voigt, C.; McGough III, R.L.; Goodman, M.; Huard, J.; Weiss, K.R. ALDH Activity Correlates with Metastatic Potential in Primary Sarcomas of Bone. JCT 2014, 05, 331–338.
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a Cancer Stem Cell Biomarker. J. Drug Target. 2019, 27, 257–269.
- Korn, P.; Kampmann, A.; Spalthoff, S.; Jehn, P.; Tavassol, F.; Lentge, F.; Gellrich, N.-C.; Zimmerer, R. Suitability of CD133 as a Marker for Cancer Stem Cells in Melanoma. Asian Pac. J. Cancer Prev 2021, 22, 1591–1597.
- Wang, K. Targeting Cancer Stem Cells by Disulfiram and Copper Sensitizes Radioresistant Chondrosarcoma to Radiation. Cancer Lett. 2021, 505, 37–48.
- Menendez, S.T.; Rey, V.; Martinez-Cruzado, L.; Gonzalez, M.V.; Morales-Molina, A.; Santos, L.; Blanco, V.; Alvarez, C.; Estupiñan, O.; Allonca, E.; et al. SOX2 Expression and Transcriptional Activity Identifies a Subpopulation of Cancer Stem Cells in Sarcoma with Prognostic Implications. Cancers 2020, 12, 964.
This entry is offline, you can click here to edit this entry!