Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Inorganic & Nuclear
Biomass wastes are produced daily by different sources, such as residues from forestry, agriculture, and food industry or animal, food, and municipal solid wastes. Bioconversion of this non-edible biomass using microorganisms recently emerged as a promising green strategy to valorize the non-starch polymers that not only plants but crustaceans as well use to support and protect themselves.
- oxidative metalloenzymes
- molecular modeling
- biopolymers
- plastic
1. Nature Strategy for Rubber Degradation: Heme Oxygenases
In Nature, several strains of NR-degrading bacteria exist whose activity solely relies on oxidative metalloenzymes [21,268,269,270,271]. Indeed, the lack of hydrolyzable groups makes polyisoprene completely inert toward hydrolytic enzymes. To date, three families of extracellular heme-enzymes, isolated from NR-degrading bacteria, with dioxygenase activity towards poly(1,4-isoprene) have been characterized, namely rubber oxygenases A (RoxA), rubber oxygenases B (RoxB), and latex clearing proteins (Lcp) (Figure 12a,b) [13,268,272]. RoxA and RoxB, active towards poly(cis-1,4-isoprene) (the most abundant polyisoprene form in nature), are distantly related from an evolutionary standpoint and are both found in Gram-negative rubber-degrading bacteria. In particular, RoxB can be considered a separate subgroup of RoxA homologs. Lcp, active towards both poly(cis-1,4-isoprene) and poly(trans-1,4-isoprene), is found, instead, in Gram-positive bacteria and does not share any sequence/structural relationship with either RoxA or RoxB [273].
The catalytic activity of RoxA, RoxB, and Lcp consists of the oxidation and breaking of the polyisoprene double bonds, forming oligoisoprenoids with terminal keto or aldehyde groups (Figure 12c). In general, these enzymes follow two distinct oxidation strategies that lead to different product distributions: RoxA catalyzes isoprene cleavage producing a C15 isoprenoid (namely 12-oxo-4,8-dimethyl-trideca-4,8-diene-1-al, ODTD) as a single major product, while RoxB and Lcp form a mixture of oligoisoprenoids of different length (C20, C25, C30, and larger) [274,275,276,277,278,279,280]. Hence, RoxA catalyzes processive oxidations starting from one end of the substrate with an exo-type action, and the pocket volume strictly governs the length of the product. RoxB and Lcp, instead, oxidize randomly NR at different locations with an endo-cleavage mode of action. Since Lcp and RoxB/A are not evolutionarily related, it means that polyisoprene-degrading pathways have evolved independently in different species.
RoxA from Xanthonomas sp. has been structurally characterized and shows an unusually low content of secondary structure elements (Figure 12a) [281]. Indeed, 67.3% of the protein structure is represented by loops, only 31.6% by helices, and the rest by a two-strended β-sheet. RoxA contains two c-type heme groups, one (heme1) in the N-terminal and one (heme2) in the C-terminal region, that are spaced by 21.4 Å and that feature different midpoint redox potentials (−65 and −130/−160 mV, respectively) [281,282]. The two heme cofactors are covalently linked to the protein via two cysteine residues each (Cys191 and Cys194; and Cys390 and Cys393, respectively) belonging to two distinct heme-binding motifs. Heme1 is the catalytic center and binds His195 as the proximal axial ligand and one dioxygen molecule as a stable distal axial ligand (as indicated by both structural and spectroscopic data). Heme2, instead, is less important for activity and His641 and His394 serve as proximal and distal axial ligands for its Fe, respectively [282].
Phe residue (Phe317) in the proximity of the coordinated O2 molecule is predicted to stabilize the binding of the latter to heme1. Mechanistic insights are scarce since no structural information on the RoxA–substrate interaction is available and eventual hydrophobic tunnel(s) for substrate access are not clearly visible in the experimentally solved structure. However, three loops are found near the distal face of heme1 that are rich in hydrophobic residues with rotationally flexible side chains that are supposed to assist NR binding within the protein matrix via substrate-induced conformational changes [281].
As for RoxB, no 3D structure is available, but it reasonably shares the same fold with RoxA and, as the latter, features two heme-binding motifs in the C- and N-terminal regions. Since RoxB has an endo-type activity, it is supposed to have a more extended binding groove with respect to RoxA, since it would facilitate random oxidation of the NR polymer [13].
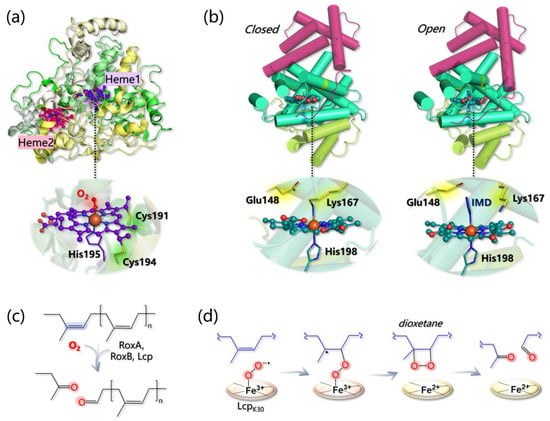
Figure 12. (a) Structure of RoxA (PDB 4B2N) and detail on heme1 coordination. (b) Closed (left) and open (right) conformations of Lcp (PDB 5O1L and 5O1M, respectively). IMD refers to the axial imidazole ligand of heme, that is found in the 3D structure of the open state. In both states, the globin core, containing the heme active site, is colored in green-cyan, while the other three- and six-helices domains are colored in light green and purple, respectively. (c) General mechanism of dioxygenation by RoxA, RoxB, and Lcp. (d) Energetically favored pathway as calculated in [283].
Lcp architecture is definitely different from the one of RoxA/B and, as indicated by the crystal structure of an Lcp from Streptomyces sp. K30 (the only one available), the protein core is characterized by a canonical globin fold formed by eight helices labeled from A to H in analogy with hemoglobin (Figure 12b) [284]. Both the N- and C-terminus are additional domains of three and six helices, respectively, with unknown function. In contrast to RoxA/B, only one type-b heme group is found in Lcp, and it is predicted to have a different coordination environment according to the conformation assumed by the whole protein. Indeed, the Lcp 3D structure has been solved in both a “closed” and an “open” conformation: in the former, Fe is axially bound to His198 and Lys167, while in the latter, Lys167 is not coordinated and the accessibility to the heme is increased. The open state has been obtained in the presence of imidazole, which, in fact, replaces Lys167 as the axial ligand in the 3D structure. The equilibrium between the two conformations has been also confirmed by EPR spectroscopy, since both low-spin (6-coordinated) and high-spin (5-coordinated) Fe centers can be detected, and the high-spin species disappears upon imidazole addition [284].
Mechanistically, it was first proposed that Glu148 in Lcp may abstract one proton from an allylic position of NR triggering the oxidation process [284]. However, this possibility has been ruled out by a subsequent computational investigation performed by Zhang and Liu at the QM/MM level [285]. The authors first modeled the Lcp-NR interaction by docking a four-unit NR substrate into the Lcp cavity (in the open state), and by refining the most reasonable and productive pose via MD simulations. Then, different catalytic pathways have been envisioned and computationally dissected, and the main results can be summarized as follows:
-
The Fe(II)-O2 system can be better described as an open shell singlet Fe(III)-O2− species, and the distal O atom of O2− is predicted to be more reactive than the proximal one, as indicated by its larger spin density;
-
Glu148 cannot act as a base since the proton abstraction from the polyisoprene allylic position is energetically prohibitive. Geometry optimizations of the Lcp-substrate system considering three models for the mutants E148A, E148Q, and E148H revealed that the distance between the O2 ligand and the double bond of the substrate (undergoing oxidation) increases, suggesting that although Glu148 is not directly involved in the reaction, it can indirectly control substrate positioning within the pocket for productive catalysis.
-
The energy profiles for assumed reaction pathways have been calculated, accounting for the different spin states that may originate along catalysis. O2− can react with either one carbon or the other forming the isoprene C=C bond and, in each case, catalysis can proceed by forming a dioxetane or an epoxide intermediate. Therefore, four pathways can be envisioned overall. The most likely one (i) involves the C carbon that is closest to the O2− ligand and (ii) entails the formation of a dioxetane intermediate, with an overall activation barrier of 15.5 kcal/mol (Figure 12d).
Remarkably, both NR and vulcanized rubber can be also oxidized by laccases and peroxidases (such as MnP) that are able to trigger the cleavage of both sulfide bridges and C=C double bonds of poly(cis-isoprene) backbone via radical chemistry according to a mechanism that has not been computationally unraveled yet [20,286,287,288].
2. Laccases and Heme Peroxidases for Plastic Degradation
Plastic pollution is an emergency experienced by all different environments throughout the planet, but it is particularly urgent in relation to the marine environment. Indeed, plastics, at variance with natural polymers, such as rubber, are materials recalcitrant to degradation and thus can have a very long half-life in the environment. For example, in the case of polyethylene terephthalate, the half-life in the ocean is estimated to be longer than 2500 years [289].
Interestingly, metalloenzymes with ligninolytic activity (i.e., laccases and peroxidases) proved to be effective in plastic biodegradation, such as PE and nylon-6,6. As for laccases, LMS was successfully employed to degrade both PE and nylon-6,6 in a pioneering contribution by Fujisawe et al. [19]: in the case of PE, its molecular weight decreased by 88.3% after 3 days of treatment with LMS and 1-hydroxybenzotriazole as a mediator. Recently, the idea that laccases are able to degrade PE is gaining ground [290,291]. In the case of the actinomycete Rhodococcus ruber, laccase incubated with PE [292] yielded a reduction of 20% of the polymer average molecular weight. More recently, the transfer of carbon atoms from 13C-labeled PE was observed [293] demonstrating that such a microorganism can use PE as a source of biomass and electrons.
Finally, laccase from Bacillus subtilis is able to increase the heavy oil biodegradation efficiency of bacterial consortia by approximately 15% [294]. An interesting aspect of the PE degradation by laccases is that the substrate binding is mostly due to hydrophobic interactions involving PE aliphatic chains, while hydrogen bonds would form with the alcohol or ketone groups of the pre-oxidized part of the substrate (if present). Moreover, during the monoelectronic oxidation process, aliphatic carbon radicals should be produced. This hypothesis was recently suggested in the context of an investigation on Rhodococcus opacus (strain R7), which produces laccase-like enzymes in the presence of PE [18,295]. Moreover, LSM with a laccase from Bacillus subtilis was employed on UV-treated low-density PE film sheets, evidencing a 40% loss of their molecular weight [18].
MnP and LiP have been reported to be involved in PE/nylon-6,6 degradation too [232,296], although their activity has been poorly characterized if compared to that of laccases. In some cases, PE and/or nylon-6,6 degradation by different bacterial cultures or fungi has been ascribed to the action of extracellular peroxidases (MnP or LiP) [17,296,297,298,299,300,301]. The activity of peroxidases toward plastics can be stimulated by H2O2, but MnP is supposed to be active towards PE even in the absence of H2O2 if a proper surfactant is employed [302].
It is still to be ascertained whether (and how) laccases/peroxidases can oxidize C-C bonds or only pre-oxidized portions of hydrocarbons. Computational tools could be suited to address this point, but the lack of structural information regarding the metalloenzyme–plastic complexes makes the building up of reliable in silico models challenging. A recent molecular docking investigation was performed in an attempt to propose possible PE biodegradation pathways by both laccases and heme-peroxidases [208]. The docking of a C12 linear alkane, used as a PE model, suggested a possible favorable binding at laccase T1 and MnP/LiP active sites (occurring with similar binding energy in all cases), although the simple substrate used is quite far from a folded and more realistic PE model [303]. In the case of laccase, the authors hypothesized that during the PE oxidation process, H2O2 is produced instead of H2O, a speculation that fits with the recent observations made by Perna et al. [304] that lignin oxidation induces enough H2O2 production to activate LPMO.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24076368
This entry is offline, you can click here to edit this entry!