ATRX is named for its causal role in ATR-X syndrome (α-thalassemia with mental impairment, X-linked), an X-linked disorder characterized by developmental delays, urogenital abnormalities, distinctive craniofacial features, and α-thalassemia caused by insufficient α-globin expression. Because of the central role of decreased α-globin mRNA expression in the ATR-X phenotype, research on ATRX initially focused on its potential as a transcriptional regulator. In fact, ATRX in concert with DAXX play wide-ranging roles in maintaining chromatin and reckoning with problematic DNA repeat sequences, with downstream effects on gene expression that have critical impacts in development. Proliferating cells must enact a telomere maintenance mechanism to ensure genomic stability. In a subset of tumors, telomeres are maintained not by telomerase, but through a homologous recombination-based mechanism termed Alternative Lengthening of Telomeres or ALT. The ALT process is linked to mutations in the ATRX/DAXX/H3.3 histone chaperone complex. This complex is responsible for depositing non-replicative histone variant H3.3 at pericentric and telomeric heterochromatin but has also been found to have roles in ameliorating replication in repeat sequences and in promoting DNA repair.
- ATRX
- DAXX
- ALT
1. Introduction
2. ATRX/DAXX and the Suppression of ALT
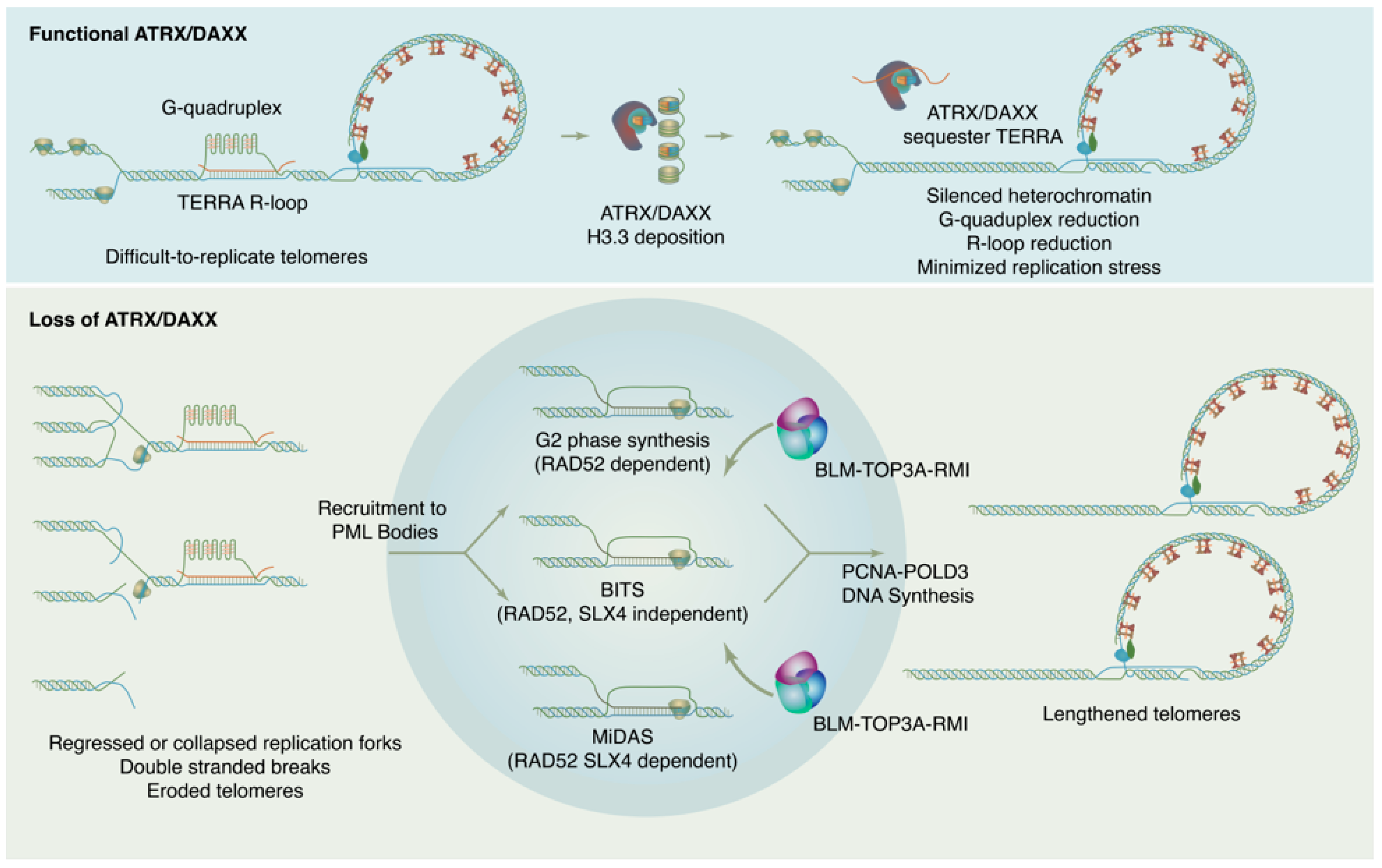
This entry is adapted from the peer-reviewed paper 10.3390/genes14040790
References
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015.
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791.
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med 1997, 3, 1271–1274.
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248.
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425.
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772.
- Bower, K.; Napier, C.E.; Cole, S.L.; Dagg, R.A.; Lau, L.M.; Duncan, E.L.; Moy, E.L.; Reddel, R.R. Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of Alternative Lengthening of Telomeres. PLoS ONE 2012, 7, e50062.
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691.
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080.
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265.
- Sieverling, L.; Hong, C.; Koser, S.D.; Ginsbach, P.; Kleinheinz, K.; Hutter, B.; Braun, D.M.; Cortes-Ciriano, I.; Xi, R.; Kabbe, R.; et al. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat. Commun. 2020, 11, 733.
- de Nonneville, A.; Reddel, R.R. Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX. Nat. Commun. 2021, 12, 1552.
- Feuerbach, L. Formal reply to “Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX”. Nat. Commun. 2021, 12, 1551.
- Wang, S.S.; Zakian, V.A. Telomere-telomere recombination provides an express pathway for telomere acquisition. Nature 1990, 345, 456–458.
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 1993, 73, 347–360.
- Biessmann, H.; Mason, J.M.; Ferry, K.; d’Hulst, M.; Valgeirsdottir, K.; Traverse, K.L.; Pardue, M.L. Addition of telomere-associated HeT DNA sequences “heals” broken chromosome ends in Drosophila. Cell 1990, 61, 663–673.
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450.
- Cives, M.; Partelli, S.; Palmirotta, R.; Lovero, D.; Mandriani, B.; Quaresmini, D.; Pelle, E.; Andreasi, V.; Castelli, P.; Strosberg, J.; et al. DAXX mutations as potential genomic markers of malignant evolution in small nonfunctioning pancreatic neuroendocrine tumors. Sci. Rep. 2019, 9, 18614.
- Singhi, A.D.; Liu, T.C.; Roncaioli, J.L.; Cao, D.; Zeh, H.J.; Zureikat, A.H.; Tsung, A.; Marsh, J.W.; Lee, K.K.; Hogg, M.E.; et al. Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2017, 23, 600–609.
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203.
- de Wilde, R.F.; Heaphy, C.M.; Maitra, A.; Meeker, A.K.; Edil, B.H.; Wolfgang, C.L.; Ellison, T.A.; Schulick, R.D.; Molenaar, I.Q.; Valk, G.D.; et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod. Pathol. 2012, 25, 1033–1039.
- Minasi, S.; Baldi, C.; Gianno, F.; Antonelli, M.; Buccoliero, A.M.; Pietsch, T.; Massimino, M.; Buttarelli, F.R. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Child’s Nerv. Syst. 2021, 37, 809–818.
- Ogino, H.; Nakabayashi, K.; Suzuki, M.; Takahashi, E.; Fujii, M.; Suzuki, T.; Ayusawa, D. Release of telomeric DNA from chromosomes in immortal human cells lacking telomerase activity. Biochem. Biophys. Res. Commun. 1998, 248, 223–227.
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179.
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londono-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731.
- Murnane, J.P.; Sabatier, L.; Marder, B.A.; Morgan, W.F. Telomere dynamics in an immortal human cell line. EMBO J. 1994, 13, 4953–4962.
- Rivera, T.; Haggblom, C.; Cosconati, S.; Karlseder, J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017, 24, 30–39.
- Mazzucco, G.; Huda, A.; Galli, M.; Piccini, D.; Giannattasio, M.; Pessina, F.; Doksani, Y. Telomere damage induces internal loops that generate telomeric circles. Nat. Commun. 2020, 11, 5297.
- Henson, J.D.; Lau, L.M.; Koch, S.; Martin La Rotta, N.; Dagg, R.A.; Reddel, R.R. The C-Circle Assay for alternative-lengthening-of-telomeres activity. Methods 2016, 114, 74–84.
- Chen, Y.A.; Shen, Y.L.; Hsia, H.Y.; Tiang, Y.P.; Sung, T.L.; Chen, L.Y. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131.
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e953.
- Loe, T.K.; Li, J.S.Z.; Zhang, Y.; Azeroglu, B.; Boddy, M.N.; Denchi, E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020, 34, 650–662.
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590.
- Zhang, J.M.; Genois, M.M.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 2021, 81, 1027–1042.e1024.
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056.
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Taylor, S.; Mitson, M.; Bachrati, C.Z.; Higgs, D.R.; Gibbons, R.J. ATRX dysfunction induces replication defects in primary mouse cells. PloS ONE 2014, 9, e92915.
- Eid, R.; Demattei, M.-V.; Episkopou, H.; Augé-Gouillou, C.; Decottignies, A.; Grandin, N.; Charbonneau, M. Genetic inactivation of ATRX leads to a decrease in the amount of telomeric cohesin and level of telomere transcription in human glioma cells. Mol. Cell Biol. 2015, 35, 2818–2830.
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558.
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280.
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174.
- Lee, S.B.; Segura-Bayona, S.; Villamor-Paya, M.; Saredi, G.; Todd, M.A.M.; Attolini, C.S.; Chang, T.Y.; Stracker, T.H.; Groth, A. Tousled-like kinases stabilize replication forks and show synthetic lethality with checkpoint and PARP inhibitors. Sci. Adv. 2018, 4, eaat4985.
- Hoang, S.M.; Kaminski, N.; Bhargava, R.; Barroso-Gonzalez, J.; Lynskey, M.L.; Garcia-Exposito, L.; Roncaioli, J.L.; Wondisford, A.R.; Wallace, C.T.; Watkins, S.C.; et al. Regulation of ALT-associated homology-directed repair by polyADP-ribosylation. Nat. Struct. Mol. Biol. 2020, 27, 1152–1164.
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58.
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell Biol 2017, 37, e00226-17.
- Ozer, O.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846.
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235.
- Sobinoff, A.P.; Allen, J.A.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919.
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827.
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801.
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236.
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londoño-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405.
- Silva, B.; Arora, R.; Bione, S.; Azzalin, C.M. TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun. 2021, 12, 3760.
- Kaminski, N.; Wondisford, A.R.; Kwon, Y.; Lynskey, M.L.; Bhargava, R.; Barroso-Gonzalez, J.; Garcia-Exposito, L.; He, B.; Xu, M.; Mellacheruvu, D.; et al. RAD51AP1 regulates ALT-HDR through chromatin-directed homeostasis of TERRA. Mol. Cell 2022, 82, 4001–4017.e4007.
- Yadav, T.; Zhang, J.M.; Ouyang, J.; Leung, W.; Simoneau, A.; Zou, L. TERRA and RAD51AP1 promote alternative lengthening of telomeres through an R- to D-loop switch. Mol. Cell 2022, 82, 3985–4000.e3984.
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538.
- Mason-Osann, E.; Dai, A.; Floro, J.; Lock, Y.J.; Reiss, M.; Gali, H.; Matschulat, A.; Labadorf, A.; Flynn, R.L. Identification of a novel gene fusion in ALT positive osteosarcoma. Oncotarget 2018, 9, 32868–32880.
- Yost, K.E.; Clatterbuck Soper, S.F.; Walker, R.L.; Pineda, M.A.; Zhu, Y.J.; Ester, C.D.; Showman, S.; Roschke, A.V.; Waterfall, J.J.; Meltzer, P.S. Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci. Rep. 2019, 9, 4544.
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252.
- Pan, X.; Drosopoulos, W.C.; Sethi, L.; Madireddy, A.; Schildkraut, C.L.; Zhang, D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. USA 2017, 114, E5940–E5949.