Atherosclerosis is an important problem in modern medicine, the keys to understanding many aspects of which are still not available to clinicians. Atherosclerosis develops as a result of a complex chain of events in which many cells of the vascular wall and peripheral blood flow are involved. Endothelial cells, which line the vascular wall in a monolayer, play an important role in vascular biology. A growing body of evidence strengthens the understanding of the multifaceted functions of endothelial cells, which not only organize the barrier between blood flow and tissues but also act as regulators of hemodynamics and play an important role in regulating the function of other cells in the vascular wall. Krüppel-like factors (KLFs) perform several biological functions in various cells of the vascular wall. The large family of KLFs in humans includes 18 members, among which KLF2 and KLF4 are at the crossroads between endothelial cell mechanobiology and immunometabolism, which play important roles in both the normal vascular wall and atherosclerosis.
- atherosclerosis
- endothelium
- metabolism
1. Regulation of Hemodynamics and Angiogenesis
2. Immunometabolism of Endothelial Cells
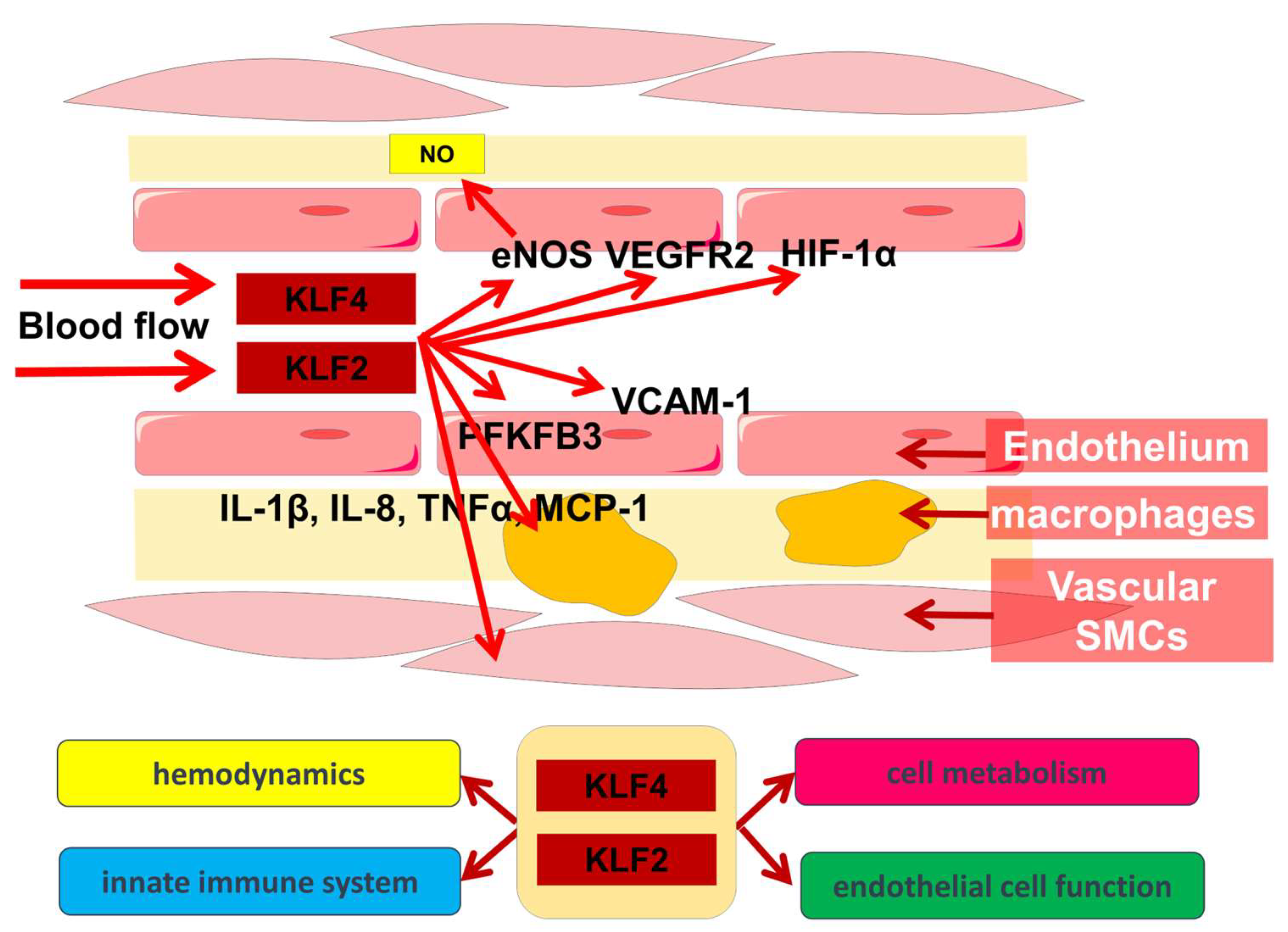
3. Participation of KLF2 and KLF4 in Other Biological Processes in the Vascular Wall
This entry is adapted from the peer-reviewed paper 10.3390/metabo13030448
References
- Nigro, P.; Abe, J.-i.; Berk, B.C. Flow Shear Stress and Atherosclerosis: A Matter of Site Specificity. Antioxid. Redox Signal. 2010, 15, 1405–1414.
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387.
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26.
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560.
- Barakat, A.I. Blood flow and arterial endothelial dysfunction: Mechanisms and implications. C. R. Phys. 2013, 14, 479–496.
- Santamaría, R.; González-Álvarez, M.; Delgado, R.; Esteban, S.; Arroyo, A.G. Remodeling of the Microvasculature: May the Blood Flow Be with You. Front. Physiol. 2020, 11, 586852.
- Tkachenko, E.; Gutierrez, E.; Saikin, S.K.; Fogelstrand, P.; Kim, C.; Groisman, A.; Ginsberg, M.H. The nucleus of endothelial cell as a sensor of blood flow direction. Biol. Open 2013, 2, 1007–1012.
- Kwon, H.B.; Wang, S.; Helker, C.S.; Rasouli, S.J.; Maischein, H.M.; Offermanns, S.; Herzog, W.; Stainier, D.Y. In vivo modulation of endothelial polarization by Apelin receptor signalling. Nat. Commun. 2016, 7, 11805.
- Wong, K.K.L.; Wu, J.; Liu, G.; Huang, W.; Ghista, D.N. Coronary arteries hemodynamics: Effect of arterial geometry on hemodynamic parameters causing atherosclerosis. Med. Biol. Eng. Comput. 2020, 58, 1831–1843.
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82.
- Porras Hernández, A.M.; Barbe, L.; Pohlit, H.; Tenje, M.; Antfolk, M. Brain microvasculature endothelial cell orientation on micropatterned hydrogels is affected by glucose level variations. Sci. Rep. 2021, 11, 19608.
- Kuhne, W.; Besselmann, M.; Noll, T.; Muhs, A.; Watanabe, H.; Piper, H.M. Disintegration of cytoskeletal structure of actin filaments in energy-depleted endothelial cells. Am. J. Physiol.—Heart Circ. Physiol. 1993, 264, H1599–H1608.
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 22–38.
- Wang, L.; Cao, Y.; Gorshkov, B.; Zhou, Y.; Yang, Q.; Xu, J.; Ma, Q.; Zhang, X.; Wang, J.; Mao, X.; et al. Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol. Res. 2019, 146, 104292.
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985.
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663.
- Dekker, R.J.; van Thienen, J.V.; Rohlena, J.; de Jager, S.C.; Elderkamp, Y.W.; Seppen, J.; de Vries, C.J.M.; Biessen, E.A.L.; van Berkel, T.J.C.; Pannekoek, H.; et al. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am. J. Pathol. 2005, 167, 609–618.
- Lee, J.S.; Yu, Q.; Shin, J.T.; Sebzda, E.; Bertozzi, C.; Chen, M.; Mericko, P.; Stadtfeld, M.; Zhou, D.; Cheng, L.; et al. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev. Cell 2006, 11, 845–857.
- Zhong, F.; Chen, H.; Wei, C.; Zhang, W.; Li, Z.; Jain, M.K.; Chuang, P.Y.; Chen, H.; Wang, Y.; Mallipattu, S.K.; et al. Reduced Krüppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int. 2015, 87, 382–395.
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145.
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquière, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48.
- Boon, R.A.; Leyen, T.A.; Fontijn, R.D.; Fledderus, J.O.; Baggen, J.M.; Volger, O.L.; van Nieuw Amerongen, G.P.; Horrevoets, A.J. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 2010, 115, 2533–2542.
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 2006, 107, 4354–4363.
- Wu, W.; Geng, P.; Zhu, J.; Li, J.; Zhang, L.; Chen, W.; Zhang, D.; Lu, Y.; Xu, X. KLF2 regulates eNOS uncoupling via Nrf2/HO-1 in endothelial cells under hypoxia and reoxygenation. Chem.-Biol. Interact. 2019, 305, 105–111.
- Kuosmanen, S.M.; Kansanen, E.; Kaikkonen, M.U.; Sihvola, V.; Pulkkinen, K.; Jyrkkänen, H.-K.; Tuoresmäki, P.; Hartikainen, J.; Hippeläinen, M.; Kokki, H.; et al. NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. 2017, 46, 1124–1138.
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220.
- Bhattacharya, R.; Senbanerjee, S.; Lin, Z.; Mir, S.; Hamik, A.; Wang, P.; Mukherjee, P.; Mukhopadhyay, D.; Jain, M.K. Inhibition of vascular permeability factor/vascular endothelial growth factor-mediated angiogenesis by the Kruppel-like factor KLF2. J. Biol. Chem. 2005, 280, 28848–28851.
- Atkins, G.B.; Jain, M.K. Role of Krüppel-Like Transcription Factors in Endothelial Biology. Circ. Res. 2007, 100, 1686–1695.
- Kawanami, D.; Mahabeleshwar, G.H.; Lin, Z.; Atkins, G.B.; Hamik, A.; Haldar, S.M.; Maemura, K.; Lamanna, J.C.; Jain, M.K. Kruppel-like factor 2 inhibits hypoxia-inducible factor 1alpha expression and function in the endothelium. J. Biol. Chem. 2009, 284, 20522–20530.
- Liu, Y.; Zheng, B.; Zhang, X.-h.; Nie, C.-j.; Li, Y.-h.; Wen, J.-k. Localization and function of KLF4 in cytoplasm of vascular smooth muscle cell. Biochem. Biophys. Res. Commun. 2013, 436, 162–168.
- Cowan, C.E.; Kohler, E.E.; Dugan, T.A.; Mirza, M.K.; Malik, A.B.; Wary, K.K. Krüppel-Like Factor-4 Transcriptionally Regulates VE-Cadherin Expression and Endothelial Barrier Function. Circ. Res. 2010, 107, 959–966.
- Lopez-Ramirez, M.A.; Lai, C.C.; Soliman, S.I.; Hale, P.; Pham, A.; Estrada, E.J.; McCurdy, S.; Girard, R.; Verma, R.; Moore, T.; et al. Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. J. Clin. Investig. 2021, 131, e139570.
- Ban, Y.; Liu, Y.; Li, Y.; Zhang, Y.; Xiao, L.; Gu, Y.; Chen, S.; Zhao, B.; Chen, C.; Wang, N. S-nitrosation impairs KLF4 activity and instigates endothelial dysfunction in pulmonary arterial hypertension. Redox Biol. 2019, 21, 101099.
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152.
- Kotlyarov, S. Immune Function of Endothelial Cells: Evolutionary Aspects, Molecular Biology and Role in Atherogenesis. Int. J. Mol. Sci. 2022, 23, 9770.
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.-F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61.
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236.
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244.
- Han, Y.; He, M.; Marin, T.; Shen, H.; Wang, W.-T.; Lee, T.-Y.; Hong, H.-C.; Jiang, Z.-L.; Garland, T.; Shyy, J.Y.J.; et al. Roles of KLF4 and AMPK in the inhibition of glycolysis by pulsatile shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2103982118.
- Chien, S. Effects of disturbed flow on endothelial cells. Ann. Biomed. Eng. 2008, 36, 554–562.
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647.
- Van Schaftingen, E.; Lederer, B.; Bartrons, R.; Hers, H.G. A kinetic study of pyrophosphate: Fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur. J. Biochem. 1982, 129, 191–195.
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018, 6, 100.
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791.
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1231–1239.
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2022, 43, 251–259.
- Xiao, W.; Loscalzo, J. Abstract 9817: Krüppel-Like Factor 4 Integrates Immunometabolism of Human Arterial Endothelial Cells. Circulation 2021, 144, A9817.
- Cargill, K.R.; Stewart, C.A.; Park, E.M.; Ramkumar, K.; Gay, C.M.; Cardnell, R.J.; Wang, Q.; Diao, L.; Shen, L.; Fan, Y.-H.; et al. Targeting MYC-enhanced glycolysis for the treatment of small cell lung cancer. Cancer Metab. 2021, 9, 33.
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. 2018, 9, 129.
- Florea, V.; Bhagavatula, N.; Simovic, G.; Macedo, F.Y.; Fock, R.A.; Rodrigues, C.O. c-Myc is essential to prevent endothelial pro-inflammatory senescent phenotype. PLoS ONE 2013, 8, e73146.
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Grechko, A.V.; Shakhpazyan, N.K.; Orekhov, A.N. The Role of KLF2 in the Regulation of Atherosclerosis Development and Potential Use of KLF2-Targeted Therapy. Biomedicines 2022, 10, 254.
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004, 199, 1305–1315.
- Di, X.; Tang, X.; Di, X. Montelukast inhibits oxidized low-density lipoproteins (ox-LDL) induced vascular endothelial attachment: An implication for the treatment of atherosclerosis. Biochem. Biophys. Res. Commun. 2017, 486, 58–62.
- Niu, N.; Xu, S.; Xu, Y.; Little, P.J.; Jin, Z.-G. Targeting Mechanosensitive Transcription Factors in Atherosclerosis. Trends Pharmacol. Sci. 2019, 40, 253–266.
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone, M.A., Jr.; et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Investig. 2006, 116, 49–58.
- Das, H.; Kumar, A.; Lin, Z.; Patino, W.D.; Hwang, P.M.; Feinberg, M.W.; Majumder, P.K.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 6653–6658.
- Atkins, G.B.; Wang, Y.; Mahabeleshwar, G.H.; Shi, H.; Gao, H.; Kawanami, D.; Natesan, V.; Lin, Z.; Simon, D.I.; Jain, M.K. Hemizygous deficiency of Krüppel-like factor 2 augments experimental atherosclerosis. Circ. Res. 2008, 103, 690–693.
- Jha, P.; Das, H. KLF2 in regulation of NF-κB-mediated immune cell function and inflammation. Int. J. Mol. Sci. 2017, 18, 2383.
- Sweet, D.R.; Vasudevan, N.T.; Fan, L.; Booth, C.E.; Keerthy, K.S.; Liao, X.; Vinayachandran, V.; Takami, Y.; Tugal, D.; Sharma, N.; et al. Myeloid Krüppel-like factor 2 is a critical regulator of metabolic inflammation. Nat. Commun. 2020, 11, 5872.
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerzsten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem. 2007, 282, 13769–13779.
- Yoshida, T.; Yamashita, M.; Horimai, C.; Hayashi, M. Deletion of Krüppel-like factor 4 in endothelial and hematopoietic cells enhances neointimal formation following vascular injury. J. Am. Heart Assoc. 2014, 3, e000622.
- Yoshida, T.; Yamashita, M.; Iwai, M.; Hayashi, M. Endothelial Krüppel-Like Factor 4 Mediates the Protective Effect of Statins against Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 1379–1388.
- Zheng, B.; Han, M.; Wen, J.-K. Role of Krüppel-like factor 4 in phenotypic switching and proliferation of vascular smooth muscle cells. IUBMB Life 2010, 62, 132–139.
- Yoshida, T.; Kaestner, K.H.; Owens, G.K. Conditional Deletion of Krüppel-Like Factor 4 Delays Downregulation of Smooth Muscle Cell Differentiation Markers but Accelerates Neointimal Formation Following Vascular Injury. Circ. Res. 2008, 102, 1548–1557.
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 2005, 280, 9719–9727.
- Castiglioni, S.; Monti, M.; Arnaboldi, L.; Canavesi, M.; Ainis Buscherini, G.; Calabresi, L.; Corsini, A.; Bellosta, S. ABCA1 and HDL3 are required to modulate smooth muscle cells phenotypic switch after cholesterol loading. Atherosclerosis 2017, 266, 8–15.
- Sharma, N.; Lu, Y.; Zhou, G.; Liao, X.; Kapil, P.; Anand, P.; Mahabeleshwar, G.H.; Stamler, J.S.; Jain, M.K. Myeloid Krüppel-Like Factor 4 Deficiency Augments Atherogenesis in ApoE−/− Mice—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2836–2838.
- Sweet, D.R.; Lam, C.; Jain, M.K. Evolutionary Protection of Krüppel-Like Factors 2 and 4 in the Development of the Mature Hemovascular System. Front. Cardiovasc. Med. 2021, 8, 645719.
- Kapoor, N.; Niu, J.; Saad, Y.; Kumar, S.; Sirakova, T.; Becerra, E.; Li, X.; Kolattukudy, P.E. Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J. Immunol. 2015, 194, 6011–6023.
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749.
- Herta, T.; Bhattacharyya, A.; Rosolowski, M.; Conrad, C.; Gurtner, C.; Gruber, A.D.; Ahnert, P.; Gutbier, B.; Frey, D.; Suttorp, N.; et al. Krueppel-Like Factor 4 Expression in Phagocytes Regulates Early Inflammatory Response and Disease Severity in Pneumococcal Pneumonia. Front. Immunol. 2021, 12, 726135.
- Oishi, Y.; Manabe, I. Krüppel-Like Factors in Metabolic Homeostasis and Cardiometabolic Disease. Front. Cardiovasc. Med. 2018, 5, 69.
- Ling, J.; Brey, C.; Schilling, M.; Lateef, F.; Lopez-Dee, Z.P.; Fernandes, K.; Thiruchelvam, K.; Wang, Y.; Chandel, K.; Rau, K.; et al. Defective lipid metabolism associated with mutation in klf-2 and klf-3: Important roles of essential dietary salts in fat storage. Nutr. Metab. 2017, 14, 22.
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 139–143.
- Lingrel, J.B.; Pilcher-Roberts, R.; Basford, J.E.; Manoharan, P.; Neumann, J.; Konaniah, E.S.; Srinivasan, R.; Bogdanov, V.Y.; Hui, D.Y. Myeloid-specific Krüppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ. Res. 2012, 110, 1294–1302.
- Li, Z.; Martin, M.; Zhang, J.; Huang, H.-Y.; Bai, L.; Zhang, J.; Kang, J.; He, M.; Li, J.; Maurya, M.R.; et al. Krüppel-Like Factor 4 Regulation of Cholesterol-25-Hydroxylase and Liver X Receptor Mitigates Atherosclerosis Susceptibility. Circulation 2017, 136, 1315–1330.
- Fang, Y.; Davies, P.F. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 979–987.
- Villarreal, G., Jr.; Zhang, Y.; Larman, H.B.; Gracia-Sancho, J.; Koo, A.; García-Cardeña, G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 391, 984–989.
- Loyer, X.; Potteaux, S.; Vion, A.C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.L.; et al. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ. Res. 2014, 114, 434–443.
- Bonauer, A.; Carmona, G.; Iwasaki, M.; Mione, M.; Koyanagi, M.; Fischer, A.; Burchfield, J.; Fox, H.; Doebele, C.; Ohtani, K.; et al. MicroRNA-92a Controls Angiogenesis and Functional Recovery of Ischemic Tissues in Mice. Science 2009, 324, 1710–1713.
- Chang, Y.-J.; Li, Y.-S.; Wu, C.-C.; Wang, K.-C.; Huang, T.-C.; Chen, Z.; Chien, S. Extracellular MicroRNA-92a Mediates Endothelial Cell–Macrophage Communication. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2492–2504.
- Niculescu, L.S.; Simionescu, N.; Sanda, G.M.; Carnuta, M.G.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. MiR-486 and miR-92a Identified in Circulating HDL Discriminate between Stable and Vulnerable Coronary Artery Disease Patients. PLoS ONE 2015, 10, e0140958.
- Liu, Y.; Li, Q.; Hosen, M.R.; Zietzer, A.; Flender, A.; Levermann, P.; Schmitz, T.; Frühwald, D.; Goody, P.; Nickenig, G.; et al. Atherosclerotic Conditions Promote the Packaging of Functional MicroRNA-92a-3p Into Endothelial Microvesicles. Circ. Res. 2019, 124, 575–587.
- Wang, Z.; Zhang, J.; Zhang, S.; Yan, S.; Wang, Z.; Wang, C.; Zhang, X. MiR-30e and miR-92a are related to atherosclerosis by targeting ABCA1. Mol. Med. Rep. 2019, 19, 3298–3304.
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550.
- Liang, G.; Wang, S.; Shao, J.; Jin, Y.-J.; Xu, L.; Yan, Y.; Günther, S.; Wang, L.; Offermanns, S. Tenascin-X Mediates Flow-Induced Suppression of EndMT and Atherosclerosis. Circ. Res. 2022, 130, 1647–1659.
- Zeng, H.; Pan, T.; Zhan, M.; Hailiwu, R.; Liu, B.; Yang, H.; Li, P. Suppression of PFKFB3-driven glycolysis restrains endothelial-to-mesenchymal transition and fibrotic response. Signal Transduct. Target. Ther. 2022, 7, 303.
- Boon, R.A.; Fledderus, J.O.; Volger, O.L.; van Wanrooij, E.J.; Pardali, E.; Weesie, F.; Kuiper, J.; Pannekoek, H.; ten Dijke, P.; Horrevoets, A.J. KLF2 suppresses TGF-beta signaling in endothelium through induction of Smad7 and inhibition of AP-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 532–539.
- Lin, Z.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A., Jr.; Balasubramanian, V.; García-Cardeña, G.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ. Res. 2005, 96, e48–e57.
- Zhou, G.; Hamik, A.; Nayak, L.; Tian, H.; Shi, H.; Lu, Y.; Sharma, N.; Liao, X.; Hale, A.; Boerboom, L.; et al. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J. Clin. Investig. 2012, 122, 4727–4731.
- Huang, J.; Pu, Y.; Zhang, H.; Xie, L.; He, L.; Zhang, C.-L.; Cheng, C.K.; Huo, Y.; Wan, S.; Chen, S.; et al. KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling. Circ. Res. 2021, 129, e87–e100.