Redox-active mediators are now appreciated as powerful molecules to regulate cellular dynamics such as viability, proliferation, migration, cell contraction, and relaxation, as well as gene expression under physiological and pathophysiological conditions. These molecules include the various reactive oxygen species (ROS), and the gasotransmitters nitric oxide (NO∙), carbon monoxide (CO), and hydrogen sulfide (H2S). For each of these molecules, direct targets have been identified which transmit the signal from the cellular redox state to a cellular response. There is a cross-regulation existing between the redox mediators and sphingolipid molecules that have a fundamental impact on a cell’s fate and organ function.
- sphingolipids
- redox
- reactive oxygen species
- nitric oxide
- hydrogen sulfide
1. Nitric Oxide (NO∙)
2. Carbon Monoxide (CO)
3. Hydrogen Disulfide
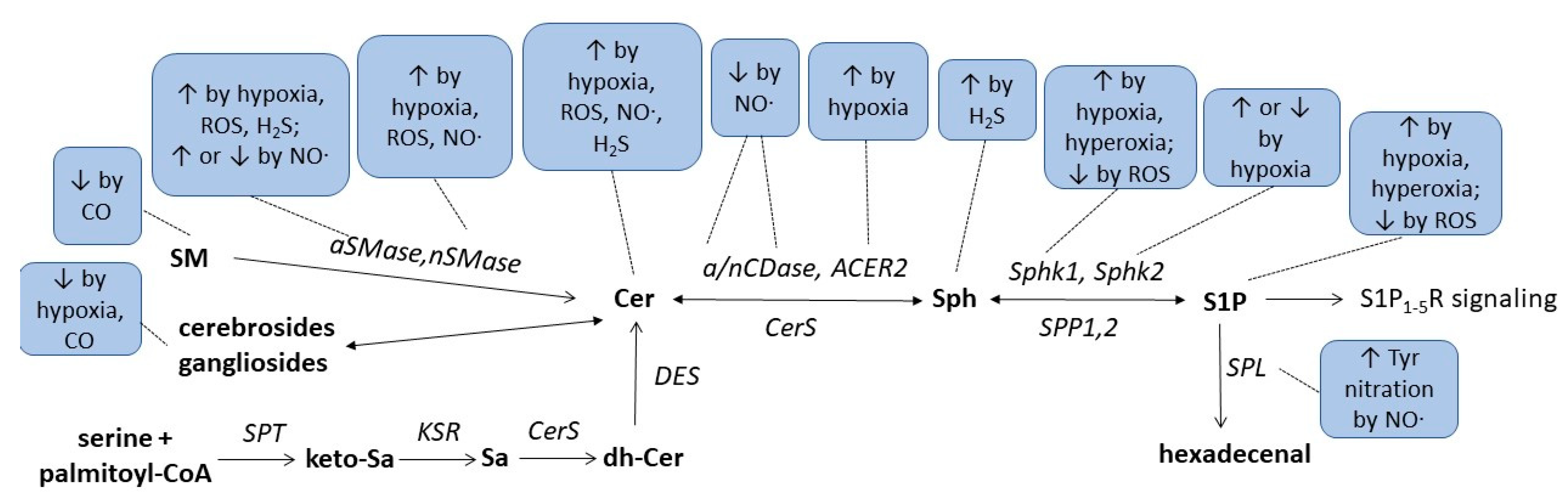
| Condition/ Enzyme |
Hypoxia | Hyperoxia | ROS | NO∙ | CO | H2S |
|---|---|---|---|---|---|---|
| Ceramides | ↑OL [44], CM [45], HC [46], PA [47] ↓VSMC [48] |
↑cancer cells [49][50][51], EC [52][53], MC [53], CM [45] |
↑MC [13][53], EC [14][53] ↓DC [16], U937 [17][18] |
↑cancer cells [41] | ||
| Sphingosine | ↑cortex [43] | |||||
| S1P | ↑VSMC [48], EC [54], cancer cells [55][56] | ↑mouse lung [57], human lung [58], EC [57] |
↓CM [59] | |||
| SM, Gangliosides | ↓brain [33][34] | |||||
| Cholesterolesters | ↑brain [33] | |||||
| Cerebrosides | ↓OL [44] | ↓brain [33] | ||||
| nSMase | ↑CM [45], PA [47] | ↑EC [52], CM [45] | ↑MC [13] | |||
| aSMase | ↑HC [46] | ↑EC [52] | ↑MC [13] ↓DC [16], U937 [17][18] |
↑cancer cells [41] | ||
| nCDase, aCDase | ↓MC [13][23] | |||||
| ACER2 | ↑adipocytes [60] | |||||
| Sphk1 | ↑EC [54], PSMC [61], cancer cells [55][62] | ↑mouse lung [57][63] | ↓CM [59] | |||
| Sphk2 | ↑cancer cells [56], PSMC [61] ↓EC [54] |
|||||
| SPL | Tyr nitration [6] | |||||
| SPT2 | ↑neuroblastoma cells [64] |
This entry is adapted from the peer-reviewed paper 10.3390/metabo13030426
References
- Huwiler, A.; Pfeilschifter, J. Nitric oxide signalling with a special focus on lipid-derived mediators. Biol. Chem. 2003, 384, 1379–1389.
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207.
- Gruetter, C.A.; Barry, B.K.; McNamara, D.B.; Gruetter, D.Y.; Kadowitz, P.J.; Ignarro, L. Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. J. Cycl. Nucleotide Res. 1979, 5, 211–224.
- Sharma, V.; Fernando, V.; Letson, J.; Walia, Y.; Zheng, X.; Fackelman, D.; Furuta, S. S-Nitrosylation in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4600.
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827.
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289.
- Lau, B.; Fazelinia, H.; Mohanty, I.; Raimo, S.; Tenopoulou, M.; Doulias, P.T.; Ischiropoulos, H. Endogenous S-nitrosocysteine proteomic inventories identify a core of proteins in heart metabolic pathways. Redox Biol. 2021, 47, 102153.
- Lander, H.M.; Ogiste, J.S.; Pearce, S.F.; Levi, R.; Novogrodsky, A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J. Biol. Chem. 1995, 270, 7017–7020.
- Pfeilschifter, J.; Huwiler, A. Nitric oxide stimulates stress-activated protein kinases in glomerular endothelial and mesangial cells. FEBS Lett. 1996, 396, 67–70.
- Callsen, D.; Pfeilschifter, J.; Brune, B. Rapid and delayed p42/p44 mitogen-activated protein kinase activation by nitric oxide: The role of cyclic GMP and tyrosine phosphatase inhibition. J. Immunol. 1998, 161, 4852–4858.
- Huwiler, A.; Pfeilschifter, J. Nitric oxide stimulates the stress-activated protein kinase p38 in rat renal mesangial cells. J. Exp. Biol. 1999, 202 Pt 6, 655–660.
- Lander, H.M.; Jacovina, A.T.; Davis, R.J.; Tauras, J.M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biol. Chem. 1996, 271, 19705–19709.
- Huwiler, A.; Pfeilschifter, J.; van den Bosch, H. Nitric oxide donors induce stress signaling via ceramide formation in rat renal mesangial cells. J. Biol. Chem. 1999, 274, 7190–7195.
- Huwiler, A.; Dorsch, S.; Briner, V.A.; van den Bosch, H.; Pfeilschifter, J. Nitric oxide stimulates chronic ceramide formation in glomerular endothelial cells. Biochem. Biophys. Res. Commun. 1999, 258, 60–65.
- Ratnayake, S.; Dias, I.H.; Lattman, E.; Griffiths, H.R. Stabilising cysteinyl thiol oxidation and nitrosation for proteomic analysis. J. Proteom. 2013, 92, 160–170.
- Falcone, S.; Perrotta, C.; De Palma, C.; Pisconti, A.; Sciorati, C.; Capobianco, A.; Rovere-Querini, P.; Manfredi, A.A.; Clementi, E. Activation of acid sphingomyelinase and its inhibition by the nitric oxide/cyclic guanosine 3′,5′-monophosphate pathway: Key events in Escherichia coli-elicited apoptosis of dendritic cells. J. Immunol. 2004, 173, 4452–4463.
- Barsacchi, R.; Perrotta, C.; Sestili, P.; Cantoni, O.; Moncada, S.; Clementi, E. Cyclic GMP-dependent inhibition of acid sphingomyelinase by nitric oxide: An early step in protection against apoptosis. Cell Death Differ. 2002, 9, 1248–1255.
- De Nadai, C.; Sestili, P.; Cantoni, O.; Lievremont, J.P.; Sciorati, C.; Barsacchi, R.; Moncada, S.; Meldolesi, J.; Clementi, E. Nitric oxide inhibits tumor necrosis factor-alpha-induced apoptosis by reducing the generation of ceramide. Proc. Natl. Acad. Sci. USA 2000, 97, 5480–5485.
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated with Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186.
- Schlossmann, J.; Desch, M. cGK substrates. Handb. Exp. Pharmacol. 2009, 191, 163–193.
- Matsumoto, A.; Comatas, K.E.; Liu, L.; Stamler, J.S. Screening for nitric oxide-dependent protein-protein interactions. Science 2003, 301, 657–661.
- Franzen, R.; Pautz, A.; Brautigam, L.; Geisslinger, G.; Pfeilschifter, J.; Huwiler, A. Interleukin-1beta induces chronic activation and de novo synthesis of neutral ceramidase in renal mesangial cells. J. Biol. Chem. 2001, 276, 35382–35389.
- Franzen, R.; Fabbro, D.; Aschrafi, A.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces degradation of the neutral ceramidase in rat renal mesangial cells and is counterregulated by protein kinase C. J. Biol. Chem. 2002, 277, 46184–46190.
- Franzen, R.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces neutral ceramidase degradation by the ubiquitin/proteasome complex in renal mesangial cell cultures. FEBS Lett. 2002, 532, 441–444.
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755.
- Ryter, S.W.; Choi, A.M. Carbon monoxide: Present and future indications for a medical gas. Korean J. Intern. Med. 2013, 28, 123–140.
- Piantadosi, C.A. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med. 2008, 45, 562–569.
- Adach, W.; Olas, B. Carbon monoxide and its donors—Their implications for medicine. Future Med. Chem. 2019, 11, 61–73.
- Kim, H.P.; Ryter, S.W.; Choi, A.M. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449.
- Brune, B.; Ullrich, V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol. Pharmacol. 1987, 32, 497–504.
- Arnold, W.P.; Aldred, R.; Murad, F. Cigarette smoke activates guanylate cyclase and increases guanosine 3′,5′-monophosphate in tissues. Science 1977, 198, 934–936.
- Graser, T.; Vedernikov, Y.P.; Li, D.S. Study on the mechanism of carbon monoxide induced endothelium-independent relaxation in porcine coronary artery and vein. Biomed. Biochim. Acta 1990, 49, 293–296.
- Wender, M. Studies of Cerebral Lipids in a Relapsing Case of Carbon Monoxide Poisoning. Acta Neuropathol. 1963, 3, 371–377.
- Mawatari, S. Biochemical study on rat brain in acute carbon monoxide poisoning. Folia Psychiatr. Neurol. Jpn. 1970, 24, 123–129.
- Weis, N.; Weigert, A.; von Knethen, A.; Brune, B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 2009, 20, 1280–1288.
- Abdelbaset-Ismail, A.; Cymer, M.; Borkowska-Rzeszotek, S.; Brzezniakiewicz-Janus, K.; Rameshwar, P.; Kakar, S.S.; Ratajczak, J.; Ratajczak, M.Z. Bioactive Phospholipids Enhance Migration and Adhesion of Human Leukemic Cells by Inhibiting Heme Oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-Dependent Manner. Stem Cell Rev. Rep. 2019, 15, 139–154.
- Jung, J.S.; Choi, M.J.; Ko, H.M.; Kim, H.S. Short-chain C2 ceramide induces heme oxygenase-1 expression by upregulating AMPK and MAPK signaling pathways in rat primary astrocytes. Neurochem. Int. 2016, 94, 39–47.
- Mitidieri, E.; Gurgone, D.; Caiazzo, E.; Tramontano, T.; Cicala, C.; Sorrentino, R.; d’Emmanuele di Villa Bianca, R. L-cysteine/cystathionine-beta-synthase-induced relaxation in mouse aorta involves a L-serine/sphingosine-1-phosphate/NO pathway. Br. J. Pharmacol. 2020, 177, 734–744.
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell Longev. 2016, 2016, 6904327.
- Whiteman, M.; Moore, P.K. Hydrogen sulfide and the vasculature: A novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell Mol. Med. 2009, 13, 488–507.
- Bae, J.; Kumazoe, M.; Yamashita, S.; Tachibana, H. Hydrogen sulphide donors selectively potentiate a green tea polyphenol EGCG-induced apoptosis of multiple myeloma cells. Sci. Rep. 2017, 7, 6665.
- Zhu, H.; Chan, K.T.; Huang, X.; Cerra, C.; Blake, S.; Trigos, A.S.; Anderson, D.; Creek, D.J.; De Souza, D.P.; Wang, X.; et al. Cystathionine-beta-synthase is essential for AKT-induced senescence and suppresses the development of gastric cancers with PI3K/AKT activation. eLife 2022, 11, e71929.
- Ni, S.J.; Yao, Z.Y.; Wei, X.; Heng, X.; Qu, S.Y.; Zhao, X.; Qi, Y.Y.; Ge, P.Y.; Xu, C.P.; Yang, N.Y.; et al. Vagus nerve stimulated by microbiota-derived hydrogen sulfide mediates the regulation of berberine on microglia in transient middle cerebral artery occlusion rats. Phytother. Res. 2022, 36, 2964–2981.
- Kendler, A.; Dawson, G. Progressive hypoxia inhibits the de novo synthesis of galactosylceramide in cultured oligodendrocytes. J. Biol. Chem. 1990, 265, 12259–12266.
- Hernandez, O.M.; Discher, D.J.; Bishopric, N.H.; Webster, K.A. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ. Res. 2000, 86, 198–204.
- Zhu, Q.; Lin, L.; Cheng, Q.; Xu, Q.; Zhang, J.; Tomlinson, S.; Jin, J.; Chen, X.; He, S. The role of acid sphingomyelinase and caspase 5 in hypoxia-induced HuR cleavage and subsequent apoptosis in hepatocytes. Biochim. Biophys. Acta 2012, 1821, 1453–1461.
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2009, 82, 296–302.
- Yun, J.K.; Kester, M. Regulatory role of sphingomyelin metabolites in hypoxia-induced vascular smooth muscle cell proliferation. Arch. Biochem. Biophys. 2002, 408, 78–86.
- Liu, B.; Hannun, Y.A. Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J. Biol. Chem. 1997, 272, 16281–16287.
- Mansat-de Mas, V.; Bezombes, C.; Quillet-Mary, A.; Bettaieb, A.; D’Orgeix, A.D.; Laurent, G.; Jaffrezou, J.P. Implication of radical oxygen species in ceramide generation, c-Jun N-terminal kinase activation and apoptosis induced by daunorubicin. Mol. Pharmacol. 1999, 56, 867–874.
- Gouaze, V.; Mirault, M.E.; Carpentier, S.; Salvayre, R.; Levade, T.; Andrieu-Abadie, N. Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol. Pharmacol. 2001, 60, 488–496.
- Huwiler, A.; Boddinghaus, B.; Pautz, A.; Dorsch, S.; Franzen, R.; Briner, V.A.; Brade, V.; Pfeilschifter, J. Superoxide potently induces ceramide formation in glomerular endothelial cells. Biochem. Biophys. Res. Commun. 2001, 284, 404–410.
- Pautz, A.; Franzen, R.; Dorsch, S.; Boddinghaus, B.; Briner, V.A.; Pfeilschifter, J.; Huwiler, A. Cross-talk between nitric oxide and superoxide determines ceramide formation and apoptosis in glomerular cells. Kidney Int. 2002, 61, 790–796.
- Schwalm, S.; Doll, F.; Romer, I.; Bubnova, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochem. Biophys. Res. Commun. 2008, 368, 1020–1025.
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. J. Biol. Chem. 2008, 283, 3365–3375.
- Schnitzer, S.E.; Welgert, A.; Zhou, J.; Brune, B. Hypoxia Enhances Sphingosine Kinase 2 Activity and Provokes Sphingosine-1-Phosphate-Mediated Chemoresistance in A549 Lung Cancer Cells. Mol. Cancer Res. 2009, 7, 393–401.
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of S1P signaling and Nox proteins. Am. J. Pathol. 2013, 183, 1169–1182.
- Ha, A.W.; Sudhadevi, T.; Ebenezer, D.L.; Fu, P.; Berdyshev, E.V.; Ackerman, S.J.; Natarajan, V.; Harijith, A. Neonatal therapy with PF543, a sphingosine kinase 1 inhibitor, ameliorates hyperoxia-induced airway remodeling in a murine model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L497–L512.
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49.
- Zhang, X.; Zhang, Y.; Wang, P.; Zhang, S.Y.; Dong, Y.; Zeng, G.; Yan, Y.; Sun, L.; Wu, Q.; Liu, H.; et al. Adipocyte Hypoxia-Inducible Factor 2alpha Suppresses Atherosclerosis by Promoting Adipose Ceramide Catabolism. Cell Metab. 2019, 30, 937–951.e5.
- Ahmad, M.; Long, J.S.; Pyne, N.J.; Pyne, S. The effect of hypoxia on lipid phosphate receptor and sphingosine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prostaglandins Other Lipid Mediat. 2006, 79, 278–286.
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642.
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L337–L351.
- Kang, M.S.; Ahn, K.H.; Kim, S.K.; Jeon, H.J.; Ji, J.E.; Choi, J.M.; Jung, K.M.; Jung, S.Y.; Kim, D.K. Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell. Signal. 2010, 22, 610–618.