Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
DNA injuries occur as a result of intrinsic or extrinsic agents and can include modifications to bases and sugars, single- and double-strand breaks (SSBs, DSBs), DNA-protein crosslinks, and base-free sites. While some specific DNA lesions can lead to mutations that cause cancer, the main consequence of DNA injuries is the threat they pose to DNA integrity and stability. To prevent accumulated DNA lesions from causing irreversible harm, cells initiate DDR, which senses the DNA damage, signals its presence, and mediates its repair.
- DNA damage response
- cancer therapy
- immunotherapy
1. Introduction
Cancer has become a leading cause of death in many countries and is still a major public health problem worldwide [1]. The classical and primary therapies are surgery, radiotherapy, and chemotherapy. Along with a better understanding of the molecular biology of the tumor cells, molecularly targeted therapies are designed to inhibit a target that is abnormal in malignant tissues when compared with normal tissues [2,3]. In comparison, most target drugs have shown limited efficacy against solid tumors, largely due to the fact that tumors frequently develop resistance to these therapies [4]. In recent years, immunotherapy has had remarkable clinical success, including immune checkpoint blockade (ICB) and adoptive cell therapy. The antibodies targeting programmed cell death 1 (PD1), PD1 ligand 1 (PDL1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) as ICBs have been approved for broad application to treat solid tumors [5]. Anti-PD therapy dominates ICB therapies and has been shown to be superior to anti-CTLA4 therapy in a wide variety of tumors [6,7]. However, the response rate of anti-PD therapy alone is usually only 20% in advanced-stage cancers, and adaptive immune resistance mechanisms also help cancer cells to escape attacks by the immune system. Thus, combining immunotherapy with other approaches to improve the anti-tumor effect is reasonable. Researchers have proposed the promising approach of utilizing DNA repair deficiency to enhance anti-tumor immunity [8].
The DNA damage response (DDR) is essential for maintaining genomic stability by repairing different types of DNA damage [9]. Cancer cells with high underlying levels of DNA damage are more dependent on DDR for survival when compared to normal cells [10]. Deficiencies in DDR result in the accumulation of DNA damage and enhance immunogenicity in tumors. Numerous studies have identified that DNA damage agents modify systemic immune functions [11,12,13]. In addition, clinical data show that a loss of mismatch repair could be a predictive biomarker for ICB response [14]. Thus, combining DDR network inhibitors with immunotherapy attracts more attention to clinical testing.
2. DNA Damage and Repair Pathway
DNA injuries occur as a result of intrinsic or extrinsic agents and can include modifications to bases and sugars, single- and double-strand breaks (SSBs, DSBs), DNA-protein crosslinks, and base-free sites [15]. While some specific DNA lesions can lead to mutations that cause cancer, the main consequence of DNA injuries is the threat they pose to DNA integrity and stability [16]. To prevent accumulated DNA lesions from causing irreversible harm, cells initiate DDR, which senses the DNA damage, signals its presence, and mediates its repair. DDR kinases, including DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated (ATM), and ataxia telangiectasia and Rad3-related (ATR), are activated at DNA lesions, which then mediate cell cycle arrest and DNA repair [17].
In the cell cycle arrest pathway, ATM and DNA-PK are mainly activated by DSBs, while ATR is activated by SSBs. These kinases phosphorylate downstream cell cycle checkpoint kinases. The active CHK1 and CHK2 then phosphorylate p53, CDC25, and WEE1, which increases the expression of p21 (p53), inhibits CDK activity and leads to cell cycle arrest at G1/S and G2/M transition (CDC25 and WEE1) [9,18]. In addition, the molecular pathways of primary DNA repair mechanisms that function in common types of DNA damage are introduced below (Figure 1) [19,20,21].
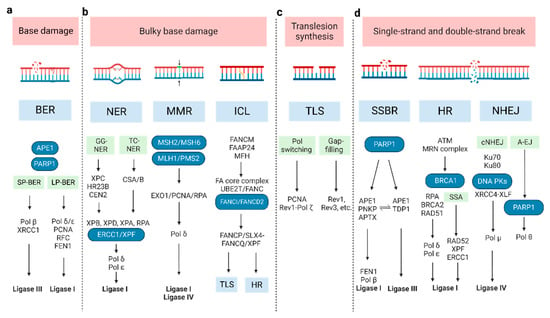
Figure 1. The molecular pathways of DNA damage repair. (a) BER can remove a single damaged base and is divided into two sub-pathways: short-patch (SP-BER) and long-patch (LP-BER). (b) For bulky base damage, NER removes the damaged base and several adjacent nucleotides, through global genome NER (GG-NER) and transcription-coupled NER (TC-NER); MMR corrects mis-incorporated bases and strand crosslinks; ICL repair, also known as FA pathway, resolves the covalently linked DNA strands. (c) TLS repair uses specialized DNA Pols to bypass DNA damage or fill single-strand DNA gaps by inserting and/or extending nucleotides, via Pol switching model and the gap-filling model. (d) SSBR repair shares most enzymatic steps with BER pathway; the main repair processes for DSB are HR, SSA, cNHEJ, and A-EJ. (A-EJ: alternative end joining; BER: base excision repair; cNHEJ: classic non-homologous end joining; DSB: double-strand break; HR: homologous recombination; ICL: inter-strand crosslink; MMR: the Mismatch Repair; NER: nucleotide excision repair; NHEJ: non-homologous end joining; SSBR: single-strand break repair; TLS: Trans lesion synthesis).
2.1. Base Damage Repair
Base excision repair (BER): Base damage occurs when chemical bonds within the DNA molecule are formed abnormally. BER can remove a single damaged base. At the beginning of BER, a series of lesion-specific DNA glycosylases remove the damaged base by cleaving the N-glycosidic bond linking the base to its corresponding deoxyribose [22]. Apurinic/apyrimidinic endonuclease 1 (APE1) and poly ADP-ribose polymerase 1 (PARP1) can sense and bind to the damage site. This catalyzes poly ADP-ribosylation (PAR) and some other protein substrates, which allows for the recruitment of repair proteins. The next synthesis/ligation step of BER is divided into two sub-pathways—short-patch and long-patch [23]. In short-patch BER, the polymerase beta (Pol β) fills the generated gap with the correct nucleotide [24]. The successive ligation of the DNA ends demands either DNA ligase I (LIG1) or the complex of DNA ligase III (LIG3) and X-ray repair cross-complementing protein 1 (XRCC1). In long-patch BER, proliferating cell nuclear antigen (PCNA), replication factor-C (RFC), flap endonuclease-1 (FEN1), Pol δ/ε, and LIG1 are included [24].
2.2. Bulky Base Damage Repair
Nucleotide excision repair (NER): This pathway removes bulky lesions, which involves removing the damaged base and several adjacent nucleotides [25]. The significant lesions initiating NER are pyrimidine dimers, such as cyclobutene pyrimidine dimers (CPD), and 6–4 photo-products induced by ultraviolet light- cisplatin-DNA intra-strand crosslinks [26,27]. In the recognition step, there are two different pathways, termed global genome NER (GG-NER) and transcription-coupled NER (TC-NER), whose recognition factor is the XPC/HR23B/CEN2 (XP complementation group C/Rad23 homolog B/Centrin-2) protein complex and CSA/B (Cockayne syndrome A and B, displacing the stalled RNA polymerase II), respectively [28,29]. The following excision and polymerization steps are all the same. XPB and XPD orchestrate the asymmetric unwinding of the DNA helix, accompanied by XPA and RPA binding to the damaged region. Then, the structure-specific endonucleases XPG and XPF/ERCC1 lead to nucleotide excision. Lastly, the resulting gap is resynthesized by Pol δ/ε and sealed by LIG1 [30].
Mismatch repair (MMR): This corrects mis-incorporated bases and strand crosslinks that occur during DNA replication. Defective MMR (dMMR) causes microsatellite instability (MSI) and an increased mutation frequency, which increases the risk of certain cancers such as Lynch syndrome and colon cancer. The MLH/MSH/PMS gene family plays a critical role in MMR [31,32]. The MSH2-MSH6 heterodimer preferentially recognizes base-base mismatches and small insertion/deletion loops (IDLs), while the MSH2-MSH3 heterodimer recognizes larger IDLs. MLH1 and PMS2, which contain the primary endonuclease activity (~90%), facilitate downstream events. The degradation of the error-containing strand is performed by Exo1 [32,33]. Then, polymerized DNA (synthesized by Pol δ), accompanied by PCNA and RPA, resynthesizes a vast gap, and LIG1 or LIG4 seals the remaining nick [34].
Inter-strand crosslink (ICL) repair: ICLs are a form of DNA damage in which two complementary DNA strands are covalently linked. To resolve ICLs, Fanconi Anemia (FA) proteins are primarily involved during the S phase of the cell cycle [35,36]. FANCM and its interacting partners (FAAP24 and MFH) recognize the lesions and recruit the FA core complex and UBE2T/FANC, to monoubiquitinate the ID2 complex (FANCI and FANCD2 heterodimer) [37,38,39]. Then, the monoubiquitinated central complex activates FANCP/SLX4-FANCQ/XPF to unhook ICLs, generating different types of lesions. These ICL-associated lesions are repaired by other DNA repair pathways, including translesion synthesis (TLS) and homologous recombination (HR) [35,40,41].
2.3. Translesion Synthesis
TLS repair: TLS, an DNA damage tolerance mechanism, uses specialized DNA Pols to bypass DNA damage or fill single-strand DNA (ssDNA) gaps by inserting and/or extending nucleotides [42]. It can be error-prone or error-free. Two models have been proposed to explain TLS: the Pol switching model and the gap-filling model [21,43]. In the former, the inserter TLS enzyme (usually a Pol h, Pol i, or Pol j), which incorporates a nucleotide opposite the DNA lesion, is replaced by extender TLS enzyme (usually Pol ζ (REV3 and REV7), in some cases by Pol j) [44,45]. The Rev1-Pol ζ complex is the most efficient among TLS Pols [44,45,46,47], initiated by monoubiquitinated PCNA [46,48]. In the latter, TLS polymerases (Rev1, Rev3, etc.) repair ssDNA to protect cells from replication stress, though the exact order of events is still unknown [49,50]. The TLS pathway has also been implicated in other DDRs, including HR, NER, and non-homologous end joining (NHEJ) [51,52,53,54].
2.4. SSB and DSB Break Repair
SSB repair (SSBR): SSBs arise either directly or indirectly (e.g., during BER of base damage) [55]. Therefore, SSBR shares several enzymatic steps with the BER pathway. In the long-patch SSBR pathway, SSBs are detected by PARP1, following end processing by APE1/PNKP (poly-nucleotide kinase 30-phosphate)/APTX (aprataxin). Next, FEN1 removes the damaged termini, following which Pol β and LIG1 repair the gap [56,57,58,59]. Different from this, APE1 recognizes the lesion and LIG3 catalyzes ligation in the short-patch SSBR pathway, while TDP1 (tyrosyl-DNA phosphodiesterase 1) executes the end-processing function in the TOP1-SSB pathway [60,61].
DSBs repair: The main processes are HR, single-strand annealing (SSA), classical NHEJ (cNHEJ), and alternative end joining (A-EJ) [62,63,64,65]. HR repair is mostly error-free [66] and only happens during the S phase and subsequent G2/M phases [67]. Firstly, the Mre11-Rad50-Nbs1 (MRN) complex senses DSBs and stably recruits ATM [68,69], which can phosphorylate itself and downstream cellular targets, including MDC1. Then, RNF8 recognizes MDC1 and promotes the ubiquitylation of histone H1 [70,71]. RNF168 recognizes ubiquitylated H1 and recruits BRCA1 and 53BP1 to mediate the HR and NHEJ pathways, respectively [72,73]. In the next step, CtIP, Exo1, and BRCA1 are implicated in the DNA end resection. The emergent ssDNA protected by replication protein A (RPA), which BRCA2 displaces, invades duplex DNA molecules through the assistance of RAD51 and BRCA1–BARD1–PALB2. With sister chromatid DNA as a template, DNA Pol δ/ε chiefly mediates the nascent strand synthesis [74,75,76], while the SSA pathway directly joins two homologous 3′ ssDNA ends after extensive DNA end resection and RPA displacement, requiring RAD52, XPF–ERCC1 and LIG1 [77,78,79].
NHEJ does not require template DNA for repair, which distinguishes it from HR. It is an error-prone means of repair which can operate throughout the cell cycle. The Ku heterodimer (Ku70 and Ku80 subunits) is needed to recognize DSB termini [80]. Then, DNA-PK is recruited by binding Ku80 [81,82]. Finally, the XRCC4-XLF, Pol μ, and LIG4 complex joins the DNA ends together to complete the damage repair [83]. When the key NHEJ components are lacking, the A-EJ pathway, also known as microhomology-mediated end joining, is enhanced in the DDR [80,84]. It requires PARP1 and Pol θ (encoded by POLQ) to elicit the re-joining of the two DNA ends by using very short homologous sequences (2–20 bp). Due to the synthetic lethal relationship between HR and the A-EJ pathway, Pol θ is a novel druggable target for cancer therapy [85,86,87].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15051619
This entry is offline, you can click here to edit this entry!