Chagas disease is a chronic systemic infection transmitted by Trypanosoma cruzi. Its life cycle consists of different stages in vector insects and host mammals. Trypanosoma cruzi strains cause different clinical manifestations of Chagas disease alongside geographic differences in morbidity and mortality. Natural killer cells provide the cytokine interferon-gamma in the initial phases of T. cruzi infection. Phagocytes secrete cytokines that promote inflammation and activation of other cells involved in defence. Dendritic cells, monocytes and macrophages modulate the adaptive immune response, and B lymphocytes activate an effective humoral immune response to T. cruzi.
- Trypanosoma cruzi
- immunity
- toll-like receptors
- virulence factors
1. Introduction
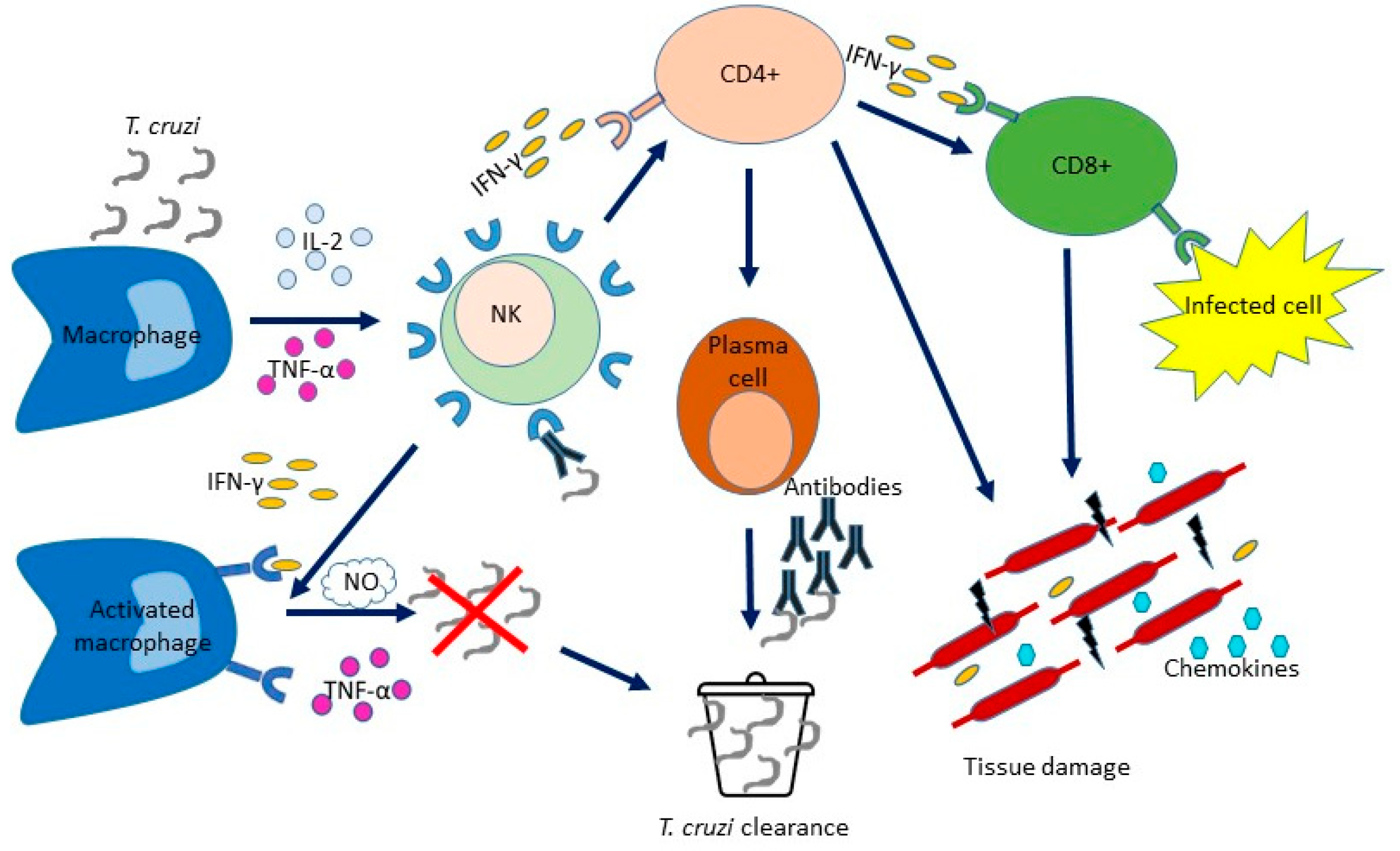
2. The Innate Immune Response to Trypanosoma cruzi
3. The Adaptative Immune Response to Trypanosoma cruzi
4. Toll-like Receptors
5. Virulence Factors
This entry is adapted from the peer-reviewed paper 10.3390/pathogens12020282
References
- Deane, L.M. Animal reservoirs of Trypanosoma cruzi in Brazil. Rev. Bras. Malariol. Doenças Trop. 1964, 16, 27–48.
- Lent, H.; Wygodzinsky, P. Revision of the Triatominae (Hemiptera Reduviidae), and their significance as vector of Chagas disease. Bull. Am. Mus. Nat. History 1979, 163, 123–520.
- Galvão, C.; Carcavallo, R.; Rocha, D.S.; Jurberg, J. A checklist of the current valid species of the subfamily Triatominae Jeannel; 1919 (Hemiptera; Reduviidae) and their geographical distribution; with nomenclatural and taxonomic notes. Zootaxa 2003, 202, 1–36.
- Schmunis, G.A. Prevention of transfusional Trypanosoma cruzi infection in Latin America. Memórias Inst. Oswaldo Cruz 1999, 94 (Suppl. 1), 93–101.
- Bern, C.; Montgomery, S.P.; Katz, L.; Caglioti, S.; Stramer, S.L. Chagas disease and the US blood supply. Curr. Opin. Infect. Dis. 2008, 21, 476–482.
- Pereira, K.S.; Schmidt, F.L.; Guaraldo, A.M.; Franco, R.M.; Dias, V.L.; Passos, L.A. Chagas disease as a foodborne illness. J. Food Prot. 2009, 72, 441–446.
- Tyler, K.M.; Engman, D.M. The life-cycle of Trypanosoma cruzi. In American Trypanosomiasis; Tyler, K.M., Miles, M.A., Eds.; World class parasites; Kluwer Academic Publishers: Boston, MA, USA, 2003; Volume 7, pp. 1–11.
- Macedo, A.M.; Machado, C.R.; Oliveira, R.P.; Pena, S.D. Trypanosoma cruzi: Genetic structure of populations and relevance of genetic variability to the pathogenesis of Chagas disease. Memórias Inst. Oswaldo Cruz 2004, 99, 1–12.
- Manoel-Caetano, F.S.; Silva, A.E. Implications of genetic variability of Trypanosoma cruzi for the pathogenesis of Chagas disease. Cad. Saúde Pública 2007, 23, 2263–2274.
- Nobrega, A.A.; Garcia, M.H.; Tatto, E.; Obara, M.T.; Costa, E.; Sobel, J.; Araujo, W.N. Oral transmission of Chagas disease by consumption of acai palm fruit; Brazil. Emerg. Infect. Dis. 2009, 15, 653–655.
- Patel, S.; Sethi, A. Imported tropical diseases. Dermatol. Ther. 2009, 22, 538–549.
- Lupi, O.; Bartlett, B.L.; Haugen, R.N.; Dy, L.C.; Sethi, A.; Klaus, S.N.; Machado Pinto, J.; Bravo, F.; Tyring, S.K. Tropical dermatology: Tropical diseases caused by protozoa. J. Am. Acad. Dermatol. 2009, 60, 897–925.
- WHO. Control of Chagas Disease; Second report of the WHO Expert Committee; Technical report series no 905; World Health Organization: Geneva, Switzerland, 2002.
- Bonney, K.M.; Engman, D.M. Chagas heart disease pathogenesis: One mechanism or many? Curr. Mol. Med. 2008, 8, 510–518.
- Nagajyothi, F.; Machado, F.S.; Burleigh, B.A.; Jelicks, L.A.; Scherer, P.E.; Mukherjee, S.; Lisanti, M.P.; Weiss, L.M.; Garg, N.J.; Tanowitz, H.B. Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell. Microbiol. 2012, 14, 634–643.
- Cardillo, F.; Postol, E.; Nihei, J.; Aroeira, L.S.; Nomizo, A.; Mengel, J. B cells modulate T cells so as to favour T helper type 1 and CD8+ T-cell responses in the acute phase of Trypanosoma cruzi infection. Immunology 2007, 122, 584–595.
- Rezende-Oliveira, K.; Sarmento, R.R.; Rodrigues, V., Jr. Production of cytokine and chemokines by human mononuclear cells and whole blood cells after infection with Trypanosoma cruzi. Rev. Soc. Bras. Med. Trop. 2012, 45, 45–50.
- Pinho, R.T.; da Silva, W.S.; de Castro Cortes, L.M.; da Silva Vasconcelos Sousa, P.; de Araujo Soares, R.O.; Alves, C.R. Production of MMP-9 and inflammatory cytokines by Trypanosoma cruzi infected macrophages. Exp. Parasitol. 2014, 147, 72–80.
- Acevedo, G.R.; Girard, M.C.; Gómez, K.A. The unsolved jigsaw puzzle of the immune response in Chagas disease. Front. Immunol. 2018, 9, 1929.
- Gurung, P.; Kanneganti, T.D. Immune responses against protozoan parasites: A focus on the emerging role of Nod-like receptors. Cell. Mol. Life Sci. 2016, 73, 3035–3051.
- Noel, W.; Raes, G.; Hassanzadeh Ghass, G.; De Baetselier, P.; Beschin, A. Alternatively activated macrophages during parasite infections. Trends. Parasitol. 2004, 20, 126–133.
- Lidani, K.C.F.; Bavia, L.; Ambrosio, A.R.; de Messias-Reason, I.J. The complement system: A prey of Trypanosoma cruzi. Front. Microbiol. 2017, 8, 607.
- Zamboni, D.S.; Lima-Junior, D.S. Inflammasomes in host response to protozoan parasites. Immunol. Rev. 2015, 265, 156–171.
- Kumar, S.; Tarleton, R.L. The relative contribution of antibody production and CD8+ T cell function to immune control of Trypanosoma cruzi. Parasite Immunol. 1998, 20, 207–216.
- Sullivan, N.L.; Eickhoff, C.S.; Sagartz, J.; Hoft, D.F. Deficiency of antigenspecific B cells results in decreased Trypanosoma cruzi systemic but not mucosal immunity due to CD8 T cell exhaustion. J. Immunol. 2015, 194, 1806–1818.
- Andrade, D.V.; Gollob, K.J.; Dutra, W.O. Acute Chagas disease: New global challenges for an old neglected disease. PLoS Negl. Trop. Dis. 2014, 8, e3010.
- Ramstead, A.G.; Robison, A.; Blackwell, A.; Jerome, M.; Freedman, B.; Lubick, K.J.; Hedges, J.F.; Jutila, M.A. Roles of Toll-Like Receptor 2 (TLR2), TLR4, and MyD88 During Pulmonary Coxiella burnetii Infection. Infect. Immun. 2016, 84, 940–949.
- Torina, A.; Blanda, V.; Villari, S.; Piazza, A.; La Russa, F.; Grippi, F.; La Manna, M.P.; Di Liberto, D.; de la Fuente, J.; Sireci, G. Immune Response to Tick-Borne Hemoparasites: Host Adaptive Immune Response Mechanisms as Potential Targets for Therapies and Vaccines. Int. J. Mol. Sci. 2020, 21, 8813.
- Sireci, G.; Badami, G.D.; Di Liberto, D.; Blanda, V.; Grippi, F.; Di Paola, L.; Guercio, A.; de la Fuente, J.; Torina, A. Recent Advances on the Innate Immune Response to Coxiella burnetii. Front. Cell. Infect. Microbiol. 2021, 11, 754455.
- Pellegrini, A.; Guiñazu, N.; Giordanengo, L.; Cano, R.C.; Gea, S. The role of Toll-like receptors and adaptive immunity in the development of protective or pathological immune response triggered by the Trypanosoma cruzi protozoan. Future Microbiol. 2011, 6, 1521–1533.
- Campos, M.A.; Gazzinelli, R.T. Trypanosoma cruzi and its components as exogenous mediators of inflammation recognized through Toll-like receptors. Mediat. Inflamm. 2004, 13, 139–143.
- Tarleton, R.L. Immune system recognition of Trypanosoma cruzi. Curr. Opin. Immunol. 2007, 19, 430–434.
- Kayama, H.; Takeda, K. The innate immune response to Trypanosoma cruzi infection. Microbes Infect. 2010, 12, 511–517.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- Carrera-Silva, E.A.; Guinazu, N.; Pellegrini, A.; Cano, R.C.; Arocena, A.; Aoki, M.P.; Gea, S. Importance of TLR2 on hepatic immune and non-immune cells to attenuate the strong inflammatory liver response during Trypanosoma cruzi acute infection. PLoS Negl. Trop. Dis. 2010, 4, e863.
- Cerbán, F.M.; Stempin, C.C.; Volpini, X.; Carrera Silva, E.A.; Gea, S.; Motran, C.C. Signaling pathways that regulate Trypanosoma cruzi infection and immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165707.
- Maganto-Garcia, E.; Punzon, C.; Terhorst, C.; Fresno, M. Rab5 activation by Toll-like receptor 2 is required for Trypanosoma cruzi internalization and replication in macrophages. Traffic 2008, 9, 1299–1315.
- Carrera-Silva, E.A.; Carolina, C.R.; Natalia, G.; Pilar, A.M.; Andrea, P.; Gea, S. TLR2, TLR4 and TLR9 are differentially modulated in liver lethally injured from BALB/c and C57BL/6 mice during Trypanosoma cruzi acute infection. Mol. Immunol. 2008, 45, 3580–3588.
- Campos, M.A.; Almeida, I.C.; Takeuchi, O.; Akira, S.; Valente, E.P.; Procópio, D.O.; Travassos, L.R.; Smith, J.A.; Golenbock, D.T.; Gazzinelli, R.T. Activation of toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J. Immunol. 2001, 167, 416–423.
- Tarleton, R.L. CD8+ T cells in Trypanosoma cruzi infection. Semin. Immunopathol. 2015, 37, 233–238.
- da Costa, T.A.; Silva, M.V.; Mendes, M.T.; Carvalho-Costa, T.M.; Batista, L.R.; Lages-Silva, E.; Rodrigues, V.; Oliveira, C.J.; Ramirez, L.E. Immunomodulation by Trypanosoma cruzi: Toward understanding the association of dendritic cells with infecting TcI and TcII populations. J. Immunol. Res. 2014, 2014, 962047.
- Van Overtvelt, L.; Vanderheyde, N.; Verhasselt, V.; Ismaili, J.; De Vos, L.; Goldman, M.; Willems, F.; Vray, B. Trypanosoma cruzi infects human dendritic cells and prevents their maturation: Inhibition of cytokines, HLA-DR, and costimulatory molecules. Infect. Immun. 1999, 67, 4033–4040.
- Sathler-Avelar, R.; Lemos, E.M.; Reis, D.D.; Medrano-Mercado, N.; Araújo-Jorge, T.C.; Antas, P.R.; Corrêa-Oliveira, R.; Teixeira-Carvalho, A.; Elói-Santos, S.M.; Favato, D.; et al. Phenotypic features of peripheral blood leucocytes during early stages of human infection with Trypanosoma cruzi. Scand. J. Immunol. 2003, 58, 655–663.
- Dutra, W.O.; Menezes, C.A.; Villani, F.N.; da Costa, G.C.; da Silveira, A.B.; Reis, D.d.; Gollob, K.J. Cellular and genetic mechanisms involved in the generation of protective and pathogenic immune responses in human Chagas disease. Memórias Inst. Oswaldo Cruz 2009, 104, 208–218.
- Souza, P.E.; Rocha, M.O.; Menezes, C.A.; Coelho, J.S.; Chaves, A.C.; Gollob, K.J.; Dutra, W.O. Trypanosoma cruzi infection induces differential modulation of costimulatory molecules and cytokines by monocytes and T cells from patients with indeterminate and cardiac Chagas’ disease. Infect. Immun. 2007, 75, 1886–1894.
- de Araújo, F.F.; Corrêa-Oliveira, R.; Rocha, M.O.; Chaves, A.T.; Fiuza, J.A.; Fares, R.C.; Ferreira, K.S.; Nunes, M.C.; Keesen, T.S.; Damasio, M.P.; et al. Foxp3+CD25(high) CD4+ regulatory T cells from indeterminate patients with Chagas disease can suppress the effector cells and cytokines and reveal altered correlations with disease severity. Immunobiology 2012, 217, 768–777.
- Ropert, C.; Ferreira, L.R.; Campos, M.A.; Procópio, D.O.; Travassos, L.R.; Ferguson, M.A.; Reis, L.F.; Teixeira, M.M.; Almeida, I.C.; Gazzinelli, R.T. Macrophage signaling by glycosylphosphatidylinositol-anchored mucin-like glycoproteins derived from Trypanosoma cruzi trypomastigotes. Microbes Infect. 2002, 4, 1015–1025.
- Oliveira, A.C.; Peixoto, J.R.; de Arruda, L.B.; Campos, M.A.; Gazzinelli, R.T.; Golenbock, D.T.; Akira, S.; Previato, J.O.; Mendonça-Previato, L.; Nobrega, A.; et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J. Immunol. 2004, 173, 5688–5696.
- Coelho, P.S.; Klein, A.; Talvani, A.; Coutinho, S.F.; Takeuchi, O.; Akira, S.; Silva, J.S.; Canizzaro, H.; Gazzinelli, R.T.; Teixeira, M.M. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes induce in vivo leukocyte recruitment dependent on MCP-1 production by IFN-gamma-primed-macrophages. J. Leukoc. Biol. 2002, 71, 837–844.
- Stahl, P.; Schwarz, R.T.; Debierre-Grockiego, F.; Meyer, T. Trypanosoma cruzi parasites fight for control of the JAK-STAT pathway by disarming their host. JAKSTAT 2015, 3, e1012964.
- Sousa-Rocha, D.; Thomaz-Tobias, M.; Diniz, L.F.A.; Souza, P.S.S.; Pinge-Filho, P.; Toledo, K.A. Trypanosoma cruzi and Its Soluble Antigens Induce NET Release by Stimulating Toll-Like Receptors. PLoS ONE 2015, 10, e0139569.
- Cronemberger-Andrade, A.; Xander, P.; Soares, R.P.; Pessoa, N.L.; Campos, M.A.; Ellis, C.C.; Grajeda, B.; Ofir-Birin, Y.; Almeida, I.C.; Regev-Rudzki, N.; et al. Trypanosoma cruzi-Infected Human Macrophages Shed Proinflammatory Extracellular Vesicles That Enhance Host-Cell Invasion via Toll-Like Receptor 2. Front. Cell. Infect. Microbiol. 2020, 10, 99.
- Castillo, C.; Muñoz, L.; Carrillo, I.; Liempi, A.; Medina, L.; Galanti, N.; Maya, J.D.; Kemmerling, U. Toll-like receptor-2 mediates local innate immune response against Trypanosoma cruzi in ex vivo infected human placental chorionic villi explants. Placenta 2017, 60, 40–46.
- Blanda, V.; Bracale, U.M.; Di Taranto, M.D.; Fortunato, G. Galectin-3 in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 9232.
- Brown, S.P.; Cornforth, D.M.; Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends. Microbiol. 2012, 20, 336–342.
- Epting, C.L.; Coates, B.M.; Engman, D.M. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp. Parasitol. 2010, 126, 283–291.
- Koo, S.-J.; Szczesny, B.; Wan, X.; Putluri, N.; Garg, N.J. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front. Immunol. 2018, 9, 202.
- Mesıas, A.C.; Garg, N.J.; Zago, M.P. Redox Balance Keepers and Possible Cell Functions Managed by Redox Homeostasis in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2019, 9, 435.
- Piacenza, L.; Peluffo, G.; Alvarez, M.N.; Martınez, A.; Radi, R. Trypanosoma cruzi antioxidant enzymes as virulence factors in chagas disease. Antioxid. Redox Signal. 2013, 19, 723–734.
- Kipnis, T.L.; David, J.R.; Alper, C.A.; Sher, A.; da Silva, W.D. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc. Natl. Acad. Sci. USA 1981, 78, 602–605.
- Norris, K.A.; Bradt, B.; Cooper, N.R.; So, M. Characterization of a Trypanosoma cruzi C3 binding protein with functional and genetic similarities to the human complement regulatory protein, decay-accelerating factor. J. Immunol. 1991, 147, 2240–2247.
- Tambourgi, D.V.; Kipnis, T.L.; da Silva, W.D.; Joiner, K.A.; Sher, A.; Heath, S.; Hall, B.F.; Ogden, G.B. A partial cDNA clone of trypomastigote decay-accelerating factor (T-DAF), a developmentally regulated complement inhibitor of Trypanosoma cruzi, has genetic and functional similarities to the human complement inhibitor DAF. Infect. Immun. 1993, 61, 3656–3663.
- Schenkman, S.; Eichinger, D.; Pereira, M.E.A.; Nussenzweig, V. Structural and functional properties of Trypanosoma trans-sialidase. Annu. Rev. Microbiol. 1994, 48, 499–523.
- Valck, C.; Ramirez, G.; Lopez, N.; Ribeiro, C.H.; Maldonado, I.; Sanchez, G.; Ferreira, V.P.; Schwaeble, W.; Ferreira, A. Molecular mechanisms involved in the inactivation of the first component of human complement by Trypanosoma cruzi calreticulin. Mol. Immunol. 2010, 47, 1516–1521.
- Chamond, N.; Gregoire, C.; Coatnoan, N.; Rougeot, C.; Freitas Junior, L.H.; da Silveira, J.F.; Degrave, W.M.; Minoprio, P. Biochemical characterization of proline racemases from the human protozoan parasite Trypanosoma cruzi and definition of putative protein signatures. J. Biol. Chem. 2003, 278, 15484–15494.
- Reina-San-Martin, B.; Degrave, W.; Rougeot, C.; Cosson, A.; Chamond, N.; Cordeiro-Da-Silva, A.; Arala-Chaves, M.; Coutinho, A.; Minoprio, P. A B-cell mitogen from a pathogenic trypanosome is a eukaryotic proline racemase. Nat. Med. 2000, 6, 890–897.
- Chamond, N.; Goytia, M.; Coatnoan, N.; Barale, J.C.; Cosson, A.; Degrave, W.M.; Minoprio, P. Trypanosoma cruzi proline racemases are involved in parasite differentiation and infectivity. Mol. Microbiol. 2005, 58, 46–60.
- Ouaissi, M.A.; Dubremetz, J.F.; Schoneck, R.; Fernandez-Gomez, R.; Gomez-Corvera, R.; Billaut-Mulot, O.; Taibi, A.; Loyens, M.; Tartar, A.; Sergheraert, C.; et al. Trypanosoma cruzi: A 52-kDa protein sharing sequence homology with glutathione S-transferase is localized in parasite organelles morphologically resembling reservosomes. Exp. Parasitol. 1995, 81, 453–461.
- Ouaissi, M.A.; Guilvard, E.; Delneste, Y.; Caron, G.; Magistrelli, G.; Herbault, N.; Thieblemont, N.; Jeannin, P. The Trypanosoma cruzi Tc52- released protein induces human dendritic cell maturation, signals via toll-like receptor 2, and confers protection against lethal infection. J. Immunol. 2002, 168, 6366–6374.
- Bonfim-Melo, A.; Ferreira, E.R.; Florentino, P.T.V.; Mortara, R.A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Front. Microbiol. 2018, 9, 1341.
- Rodrigues, A.A.; Clemente, T.M.; Dos Santos, M.A.; Machado, F.C.; Gomes, R.G.; Moreira, H.H.; Cruz, M.C.; Brígido, P.C.; Dos Santos, P.C.; Martins, F.A.; et al. A Recombinant Protein Based on Trypanosoma cruzi P21 Enhances Phagocytosis. PLoS ONE 2012, 7, e51384.
- Ferreira, É.R.; Horjales, E.; Bonfim-Melo, A.; Cortez, C.; da Silva, C.V.; De Groote, M.; Sobreira, T.J.P.; Cruz, M.C.; Lima, F.M.; Cordero, E.M.; et al. Unique behavior of Trypanosoma cruzi mevalonate kinase: A conserved glycosomal enzyme involved in host cell invasion and signaling. Sci. Rep. 2016, 6, 24610.
- de Castro Neto, A.L.; da Silveira, J.F.; Mortara, R.A. Comparative Analysis of Virulence Mechanisms of Trypanosomatids Pathogenic to Humans. Front. Cell. Infect. Microbiol. 2021, 16, 669079.
- Freire-De-Lima, L.; Fonseca, L.M.; Oeltmann, T.; Mendoncą-Previato, L.; Previato, J.O. The trans-sialidase, the major Trypanosoma cruzi virulence factor: Three decades of studies. Glycobiology 2015, 25, 1142–1149.