Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Angiotensin II (AngII)-regulated Long non-coding RNAs (lncRNAs) Alivec functions, at least in part, to mediate the AngII-induced chondrogenic transformation of vascular smooth muscle cells (VSMCs) implicated in vascular dysfunction and hypertension.
- Angiotensin II
- lncRNAs
- cardiovascular disease
- vascular smooth muscle cells
- chondrocytes
- hypertension
1. Introduction
Cardiovascular diseases (CVDs), such as hypertension and atherosclerosis, are leading causes of morbidity and mortality worldwide [1]. Vascular smooth muscle cells (VSMCs) in the arterial wall maintain vascular tone and blood pressure and are under the control of the renin–angiotensin system (RAS)-Angiotensin II (AngII) system.
In CVD or vascular injury, dysregulated growth factor and AngII signaling promotes VSMCs to switch from a contractile to synthetic phenotype [2]. The synthetic phenotype manifests in increased VSMC proliferation, hypertrophy, migration, inflammation and the key processes associated with the pathogenesis of arterial stenosis/restenosis, hypertension and atherosclerosis [3][4][5][6]. Furthermore, the synthetic VSMCs tend to transform into chondrocyte-like cells, which promotes extracellular calcium deposition and vascular dysfunction associated with these pathologies [7][8][9]. Aggrecan (Acan) is an extracellular matrix protein that is prominent in chondrocytes during cartilage formation and is upregulated in aortic VSMCs after injury [9]. The transcription factor (TF) Sox9, which regulates chondrogenesis, is associated with VSMC synthetic/chondrocyte phenotype and promotes extra-cellular matrix (ECM) alterations and calcium deposition [10]. However, the mechanisms involved in AngII-mediated phenotypic transformation of VSMC to chondrocyte-like cells are not well understood.
Long non-coding RNAs (lncRNAs) are a group of non-coding RNAs (ncRNAs) that are more than 200 nucleotides in size and are processed like protein coding mRNAs but lack protein-coding potential [11]. LncRNAs have diverse functions and regulate gene expression at the level of transcription through the interaction with and recruitment of TFs, chromatin modifier proteins and ribonucleoproteins to specific target gene loci, or via the post-transcriptional regulation of microRNAs and signaling proteins [12]. Genome-wide association studies (GWAS) identified several single nucleotide polymorphisms (SNPs) associated with CVDs that reside in the lncRNA loci [13]. LncRNAs regulate various physiological and pathological processes [14]. In VSMCs they regulate cell proliferation, migration, reactive oxygen species (ROS) production and inflammation, key factors associated with CVDs [15][16]. Researchers identified the first lncRNAs regulated by AngII in rat VSMCs (RVSMCs) using integrated analysis of RNA-seq data with ChIP-seq datasets from histone H3K4me3 and H3K36me3 profiling [17]. Since then, several VSMC lncRNAs such as SENCR, MYOSLID and SMILR were described and found to play key roles in CVDs [18][19][20].
2. Alivec Is an AngII-Induced lncRNA Adjacent to Chondrogenic Gene Acan in RVSMCs
Here analyzed RNA-seq data previously generated in a laboratory from RVSMCs treated with AngII (100 nM, 3 h) [17] using STAR aligner and observed that a previously identified novel lncRNA (lnc Ang26), which named Alivec, was highly induced by AngII (Figure 1A). To further characterize the Alivec locus, researchers integrated the RNA-seq data with histone H3K27ac (enhancer mark) ChIP-seq data from AngII treated RVSMCs [21]. Combined RNA-seq and ChIP-seq data showed that the lncRNA Alivec locus overlaps with an AngII-induced H3K27ac enriched region (Figure 1B). Alivec has 3 exons and the gene is located on rat chromosome 1 adjacent (117 kb distance) to the protein-coding gene Acan (Figure 1B). RNA-seq analyses also showed that the expression of the nearby gene Acan, was likewise increased by AngII. Furthermore, RT-qPCR validation showed that RVSMCs exposed to AngII displayed marked induction of Alivec expression (up to 30-fold) within 3 h of treatment; this persisted even at 6 h compared to the control cells (Figure 1C). Under the same conditions, the induction of Acan was also observed (Figure 1D), suggesting a potential role for Alivec in the regulation of Acan expression by AngII. This was interesting, as Acan codes for the protein aggrecan, which is known to be induced by growth factors and cytokines and is also a key biomarker of chondrogenesis associated with VSMC dysfunction in CVDs [22].
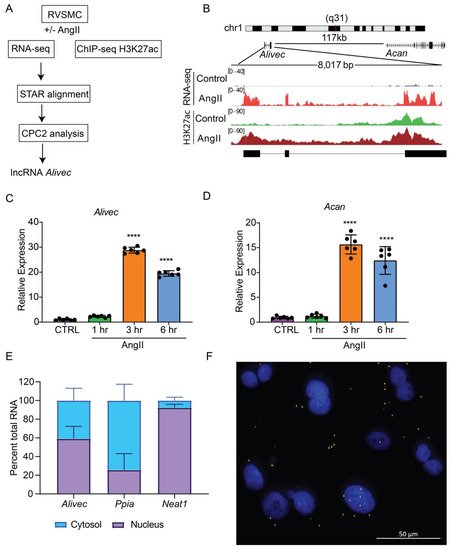
Figure 1. Alivec is an AngII-induced enhancer-associated lncRNA adjacent to chondrogenic gene Acan in RVSMCs. (A) Schematic diagram depicting RNA-seq and H3K27ac ChIP-seq alignment pipeline for the identification of lncRNA Alivec (AngII-induced lncRNA in vascular smooth muscle cells eliciting chondrogenic phenotype) exons, overlapping H3 lysine 27 acetylation (H3K27ac) enrichment and Alivec’s coding potential, which was determined using the software CPC2 (coding potential calculator 2). (B) Schematic showing genomic organization of Alivec and the neighboring gene Acan in the rat genome. Integrative Genomics Viewer (IGV) tracks showing Alivec locus with representative RNA-seq tracks (RNA-Seq) and H3K27ac ChIP-seq tracks (H3K27ac) from control- and AngII-treated RVSMCs. (C,D) RT-qPCR analysis of Alivec and Acan expression in RVSMCs treated ± AngII (100 nM) for the indicated time periods. Data presented as mean ± SD, n = 6 biological replicates, one-way ANOVA followed by Dunnett’s multiple comparisons test and **** p < 0.0001 vs. control untreated cells (CTRL (E) RT-qPCR analysis of Alivec, Ppia and Neat1 showing their relative enrichment in cytosolic and nuclear fractions of AngII-treated RVSMCs. (F) Subcellular localization of Alivec in AngII-treated RVSMCs, determined by RNA–FISH analysis. Alivec is shown as distinct yellow spots and nuclei are stained with DAPI (blue). Scale bar 50 µm.
Next, researchers performed experiments to further characterize Alivec. Rapid amplification of cDNA end (RACE)-PCR experiments verified the 5′ and 3′ ends of Alivec and defined the total transcript size to be 2275 nucleotides. Considering the localization of lncRNAs in the nucleus or cytoplasm can determine their functions, [23] researchers examined the cellular localization of lncRNA Alivec. In AngII-treated RVSMCs, sub-cellular fractionation followed by RT-qPCR showed that Alivec is distributed in the nucleus and cytosol (Figure 1E). Ppia and a lncRNA Neat1 served as controls for cytoplasmic and nuclear fractions, respectively (Figure 1E). RNA–FISH experiments with branched DNA probes, further confirming nuclear and cytoplasmic localization of Alivec, as indicated by the presence of distinct spots/foci distributed in both compartments (Figure 1F). These spots were not visible in the absence of the probes. The protein-coding potential analysis of Alivec (coding potential calculator version 2.0, CPC2) showed that it had a coding probability of 0.31, classifying it as a non-coding transcript. The lack of coding potential was confirmed by in vitro transcription/translation assays using pcDNA Alivec plasmids, which showed no detectable peptide product from Alivec, as compared to the positive luciferase control. Together, these results indicate that Alivec is an AngII-induced lncRNA in RVSMCs.
3. AngII-induces Alivec and Acan Expression via Activation of AT1R and Src Kinase
AngII, through the activation of AT1R, induces multiple signaling pathways to regulate downstream gene expression and alter VSMC function [24]. To elucidate the role of AT1R signaling in regulating Alivec and Acan expression, researchers pre-treated RVSMCs with the AT1R antagonist, losartan (10 µM, 30 min), followed by AngII (100 nM). Losartan abrogated the AngII-mediated induction of Alivec (Figure 2A). Acan expression was also attenuated by losartan (Figure 2B). Pre-treatment of RVSMCs for 30 min with specific inhibitors of key signaling kinases, ERK1/2 (U0126, 10 µM) and Src (PP1,10 µM), significantly reduced AngII-induced Alivec expression. Conversely, inhibitors of the p38 MAP kinase (SB202190, 5 µM) and JAK (Inhibitor I, 10 µM), had minimal effect on Alivec expression (Figure 2C). Interestingly, the AngII-mediated induction of Acan was significantly suppressed by inhibition of the Src kinase, and to a lesser extent, by inhibitors of ERK1/2 and JAK (Figure 2D). These results demonstrate key roles of AT1R signaling and downstream kinases in the regulation of Alivec and Acan in RVSMCs.
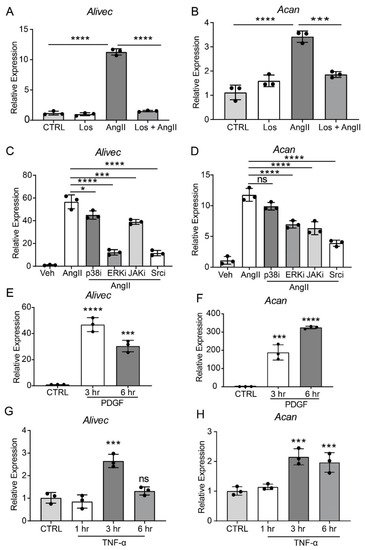
Figure 2. AngII-induced Alivec expression is regulated by AT1R and downstream kinases Src and ERK1/2. (A,B) RT-qPCR analysis of Alivec and Acan expression in RVSMCs pre-treated with the AT1R inhibitor Losartan (Los, 10 µM) for 30 min, followed by AngII treatment (100 nM, 3 h). (C,D) RVSMCs were pre-treated with vehicle DMSO (Veh) or inhibitors (i) of p38, ERK1/2, JAK and Src kinases for 30 min, followed by AngII treatment (100 nM, 3 h). (E–H) RT-qPCR analysis of Alivec and Acan expression in RVSMCs, treated with PDGF (10 ng/mL) and TNF-α (10 ng/mL). Data presented as mean ± SD. Comparisons were performed by one-way ANOVA with Tukey’s post-hoc test. (A–D) Dunnett’s multiple comparisons test (E–H), * p < 0.05, *** p < 0.001 and **** p < 0.0001 vs. CTRL or AngII.
It also was determined if Alivec is induced by other VSMC growth factors, such as PDGF, and the inflammatory cytokine TNF-α. RVSMCs treated with PDGF (10 ng/mL) and TNF-α (10 ng/ml, 3 and 6 h) showed increased Alivec and Acan mRNA levels, although with varying kinetics (Figure 2E–H) suggesting a general effect. Together, these results show that Alivec and Acan are upregulated by not only AngII, but by other growth factors and inflammatory cytokines that might co-operate to augment VSMC dysfunction upon vascular injury.
4. Alivec Knockdown Attenuates AngII-Induced Upregulation of Genes Associated with Chondrogenesis
AngII regulates the expression of inflammatory, fibrotic and calcification-related genes in VSMC [24]. To further characterize the role of Alivec in the regulation of AngII-induced gene expression in RVSMCs, a locked nucleic acid (LNA)-modified anti-sense oligonucleotide (ASO) GapmeRs-mediated knockdown of Alivec was performed. Three GapmeRs targeting Alivec were tested to assess their efficacy in RVSMCs (Supplementary Figure S2A). Results showed that GapmeR3 (denoted as AlivecGap) achieved maximum reduction (~60%) in AngII-induced Alivec expression, as compared to the control GapmeR (NCGap) (Figure 3A and Supplementary Figure S2B). RVSMCs were transfected with AlivecGap or NCGap and treated with or without AngII. RNA extracted from these cells was subjected to microarray expression profiling (Supplementary Figure S3A,B). After Alivec knockdown, researchers identified 1169 differentially expressed genes in untreated RVSMCs (676 downregulated and 493 upregulated), and 1294 differentially expressed genes in AngII-treated RVSMCs (664 downregulated and 630 upregulated), which included several chondrogenic genes (Figure 3B). Gene ontology (GO) analysis of downregulated genes showed enrichment of biological processes, such as cell adhesion and the circulatory system (Figure 3C), which are important functions of VSMC and the cardiovascular system. The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed enrichment of pathways involved in mucin type O-glycan biosynthesis, nitric oxide second messenger cGMP signaling and vascular smooth muscle contraction (Figure 3D) that could be associated with VSMC functions and hypertension.
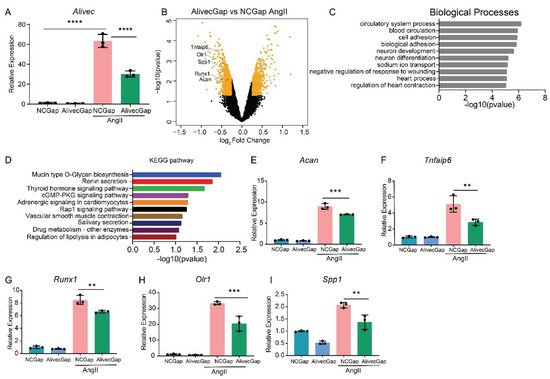
Figure 3. Alivec knockdown attenuates upregulation of AngII-induced chondrogenic genes in RVSMCs. (A) Knockdown efficiency of LNA GapmeR targeting Alivec (AlivecGap) (100 nM) vs. non-targeting control GapmeR (NCGap) (100 nM), as determined by RT-qPCR. Data presented as mean ± SD, one-way ANOVA followed by Tukey’s post-hoc test and **** p < 0.0001 vs. indicated groups. (B) Volcano plot showing differentially expressed genes (orange color) in AngII-treated RVSMCs transfected with AlivecGap vs. NCGap. Labeled dots indicate genes involved in chondrogenesis. (C) Gene ontology (GO) analysis by the TOPPGENE tool of differentially-expressed (DE) genes showing the top 10 biological processes enriched in downregulated genes after Alivec knockdown. (D) KEGG pathway analysis of differentially-expressed (DE) genes, showing the top 10 molecular pathways affected in downregulated genes after Alivec knockdown. (E–I) RT-qPCR validation of indicated chondrogenic genes after Alivec knockdown in RVSMCs treated ± AngII (100 nM, 3 h). Data presented as mean ± SD, one-way ANOVA followed by Tukey’s post-hoc test and ** p < 0.01 and *** p < 0.001 vs. indicated groups) n = 3 biological replicates.
RT-qPCR validation of microarray data confirmed downregulation of Acan and several other chondrogenic genes, including Tnfaip6, Runx1, Olr1 and Spp1 (Figure 3E–I), after Alivec knockdown in RVSMCs. In addition, Acan downregulation is consistent with the known role of lncRNAs in regulating adjacent genes (Figure 3B).
Conversely, in gain-of-function experiments, transient overexpression of Alivec increased mRNA levels of Acan, Runx1, Tnfaip6, Olr1 and Runx2, relative to the controls (Figure 4A–F). Together, these results demonstrate that lncRNA Alivec plays a key role in the regulation of AngII-induced chondrogenic genes in RVSMCs.
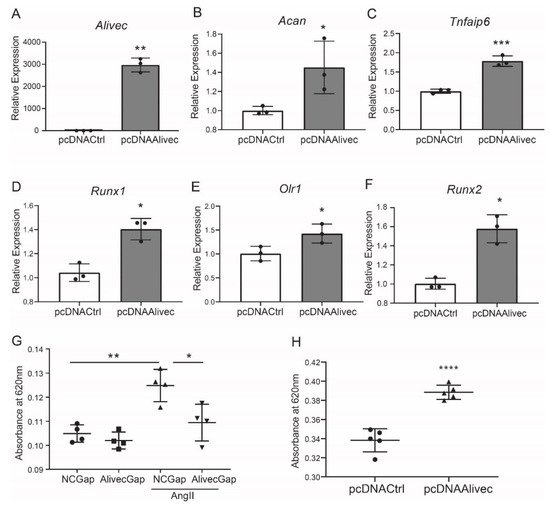
Figure 4. Alivec overexpression promotes and its knockdown inhibits the chondrogenic/osteogenic phenotype in RVSMCs. (A) RT-qPCR analysis showing expression of Alivec after transfection of RVSMC with pcDNAAlivec vs. empty vector (pcDNACtrl). (B–F) RT-qPCR analysis showing expression of target genes Acan, Tnfaip6, Runx1, Olr1 and Spp1 after overexpression of Alivec in RVSMCs. Data presented as mean ± SD, n = 3 biological replicates, unpaired two-tailed Student’s t-test and * p < 0.05, ** p < 0.01, *** p < 0.001 vs. pcDNACtrl. (G) Alcian blue staining performed on RVSMCs transfected with NCGap and AlivecGap and treated ± AngII (100 nM). Data were presented as mean ± SD, n = 4 biological replicates, one-way ANOVA followed by Tukey’s post-hoc correction and * p < 0.05, ** p < 0.01 vs. indicated groups. (H). Alcian blue staining after overexpression of Alivec in RVSMCs. Data presented as mean ± SD, n = 5 biological replicates, unpaired two-tailed Student’s t-test and **** p < 0.0001 vs. pcDNACtrl.
5. Alivec Mediates a Chondrogenic/Osteogenic Phenotype in RVSMCs
Alcian blue stains glycosaminoglycan proteins, including aggrecan, that are associated with the ECM and chondrogenic differentiation [25]. Relative to control, AngII-treated RVSMCs showed increased alcian blue staining, and this was significantly decreased by Alivec knockdown with GapmeR (Figure 4G). Conversely, overexpression of Alivec increased alcian blue staining (Figure 4H). These data demonstrate that Alivec regulates expression of several AngII-induced chondrogenic genes, including nearby Acan, and promotes a chondrocyte phenotype in RVSMCs.
6. Transcription Factor Sox9 Controls Alivec Expression in RVSMCs
Transcription factor (TF) motif analyses of 500 bases upstream of the Alivec transcription start site (TSS) showed enrichment of ten TFs, including Sox9 (Figure 5A). Sox9 regulates chondrogenesis and osteogenesis in mesenchymal stem cells [26]. researchers examined Sox9 interaction with the Alivec promoter in RVSMCs transfected with a Sox9 expression plasmid (pcDNASox9) and a control vector (pcDNACtrl), using chromatin immunoprecipitation (ChIP) assays with the Sox9 antibody. ChIP-qPCR showed enrichment of Sox9 in the predicted Sox9-binding region, upstream of the Alivec TSS, as compared with the control pcDNACtrl plasmid-transfected cells (Figure 5B). Transfection of RVSMCs with the siRNAs targeting Sox9 (siSox9), reduced the Sox9 protein and transcript levels in control- and AngII-treated cells (Figure 5C,D). Sox9 knockdown also decreased the AngII-induced expression of Alivec and Acan (Figure 5E,F). Conversely, the overexpression of Sox9 using the pcDNASox9 plasmid in RVSMCs increased Alivec and Acan vs. the control vector-transfected cells (Figure 5G–I). These results demonstrate that Sox9 can regulate Alivec and Acan expression in response to AngII in RVSMCs.
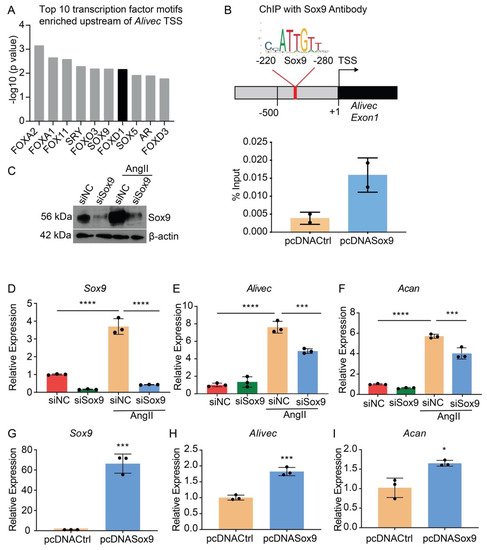
Figure 5. Transcription factor Sox9 controls Alivec expression in RVSMCs (A). Top 10 transcription factor (TF) binding motifs, enriched in the genomic region upstream of Alivec transcription start site (TSS). (B) ChIP assays with Sox9. Upper panel depicts schematic of the predicted Sox9-binding site upstream of Alivec TSS (arrow). Lower panel show ChIP-qPCR data with Sox9 antibody in RVSMCs transfected with pcDNACtrl and pcDNASox9 plasmids. ChIP-DNA was analyzed by qPCR with Alivec promoter primers overlapping Sox9-binding sites (n = 2). (C) Sox9 knockdown with siRNAs. Sox9 protein levels determined by Western blotting in RVSMCs transfected with siRNA targeting Sox9 (siSox9) or negative control (siNC) oligonucleotides, and treated ± AngII (upper panel). β-actin protein levels (lower panel) were used as internal control. (D–F) RT-qPCR analysis of indicated genes in siSox9- and siNC-transfected RVSMCs at the basal level and after stimulation with AngII. Data presented as mean ± SD, n = 3 biological replicates, one-way ANOVA followed by Tukey’s post-hoc test and *** p < 0.001, **** p < 0.0001 vs. indicated groups. (G–I). RT-qPCR analysis of indicated genes in RVSMCs, transfected with Sox9 expression plasmid (pcDNASox9) and pcDNACtrl control plasmid. Data represented as mean ± SD, n = 3 biological replicates, unpaired Student’s t-test and * p < 0.05, *** p < 0.001 vs. control plasmid.
7. Alivec RNA Interacts with hnRNPA2B1 as well as with Tropomyosin alpha-3 Chain, a Protein with Putative Association with the Contractile Phenotype of RVSMCs
LncRNAs can regulate transcription, gene expression and cellular phenotype through interactions with proteins [27][28]. Researchers performed RNA-pulldown assays with Alivec, followed by mass spectrometry, and found a number of proteins associated with Alivec, relative to negative control. STRING analysis demonstrated that the Alivec interacting proteins were associated with VSMC contractile functions, nuclear membrane organization and regulation of gene expression (Figure 6A). One of these proteins, a tropomyosin alpha-3 chain (Tpm3) [29] was noteworthy, due to the known roles of alpha-tropomyosin isoforms in VSMC contractile functions and gene regulation [30][31]. RNA–protein interaction prediction (RPISeq) software showed that the Alivec-Tpm3 RNA–protein interaction had a positive interaction probability of 0.75 (>0.5 considered positive). Researchers then performed RNA-pulldown, followed by Western blot analysis, in order to validate the Tpm3 association with Alivec (Figure 6B), which confirmed the mass spectrometry results. Specific interaction of Alivec with Tpm3 was also supported by RNA-immunoprecipitation (RNA-IP), using an antibody against Tpm3. No interaction was seen with Gapdh mRNA and H19 lncRNA (negative controls, Figure 6C). In addition, mass spectrometry showed that Alivec interacts with the RNA-binding protein, heterogeneous nuclear ribonucleoproteinA2B1 (hnRNPA2B1), which was validated by RNA-pulldown, followed by Western blotting (Figure 6B, lower panel). These results indicate that lncRNA Alivec mediates AngII-induced VSMC dysfunction via interaction with both cytoplasmic and nuclear proteins.
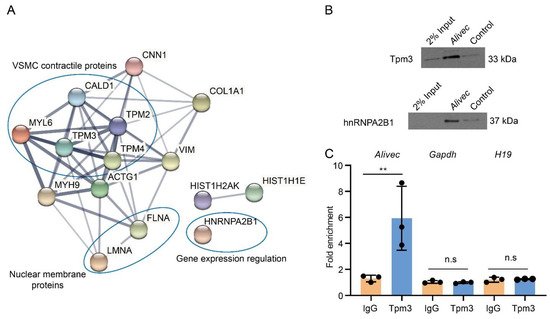
Figure 6. Alivec RNA interacts with hnRNPA2B1 and tropomyosin-3-alpha, a protein potentially associated with the contractile phenotype of VSMCs. (A) Network of protein complexes generated (using STRING database) from Alivec-specific interacting proteins identified by RNA-pulldown coupled to mass spectrometry. (B) Western blot analysis with Tpm3 antibody (upper panel) or hnRNPA2B1 antibody (lower panel) following RNA-pulldown assays with RVSMCs extracts using biotinylated Alivec RNA and poly A RNA as negative control (Control). (C) RNA immunoprecipitation assays with UV-cross-linked RVSMC cell extracts using anti-Tpm3 antibody and IgG as negative control. RNA from Tpm3 and IgG immunoprecipitates were analyzed by RT-qPCR, using indicated primers. Results were shown as fold enrichment over IgG. Data presented as mean ± SD, n = 3 biological replicates and ** p < 0.01 vs. IgG, using unpaired Student’s t-test. N.s. indicates not significant.
8. AngII Treatment Increases Aortic Expression of Alivec in Rats
Next, study examined whether AngII upregulates the expression of Alivec and Acan in vivo in rats. Male Sprague–Dawley rats (12-weeks-old), infused with AngII (200 ng/kg/min, four weeks), showed the expected increase in systolic blood pressure (SBP) compared to control vehicle (PBS) infused rats (Figure 7A). Aortic thickening was noted in AngII-infused animals relative to the controls (Figure 7B). Immunohistochemical staining showed marked increases in aggrecan and Runx1 proteins and decreases in the smooth muscle contractile proteins α-SMA and SM22 alpha (Figure 7B). Furthermore, mRNA levels of Alivec, Acan and Runx1 were significantly increased in vessels from the AngII group (Figure 7C–E).
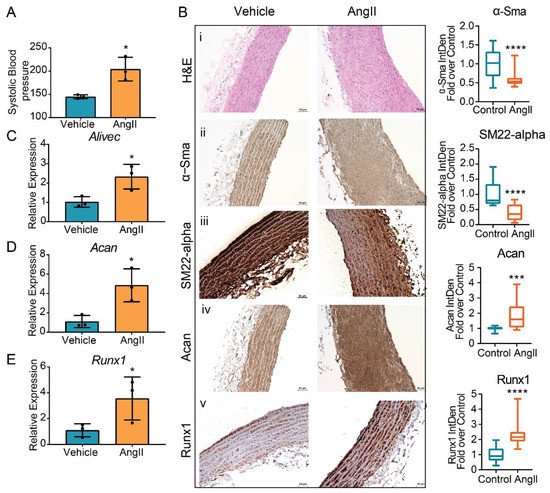
Figure 7. Regulation of Alivec and Acan in the aortas from a rat model of AngII-induced hypertension. (A) Systolic blood pressure (SBP) measured in male Sprague–Dawley rats infused with AngII or vehicle for 4 weeks. (B) Representative images of hematoxylin and eosin (H&E) staining (i) and IHC staining for α-Sma (ii), SM22-alpha (iii), Acan (iv) and Runx1 (v) proteins on aortic tissue sections from vehicle or AngII-infused rats, scale bar: 50 µM. Box plots on the right show the quantification of aortic staining of indicated proteins shown in panels (ii) to (v). Box plots on the right show integrated density (IntDen) expressed as fold-over control. Staining was quantified using ImageJ software in 20 different areas for each group (3 aortas in control and 3 in AngII group. Data represented as mean and minimum/maximum, unpaired Student’s t-test and *** p < 0.001 and **** p < 0.0001). (C–E) RT-qPCR analysis showing gene expression of Alivec, Acan and Runx1 in aortas from AngII-infused rats in comparison to vehicle-treated rats. Data presented as mean ± SD, n = 3 biologic replicates and unpaired Student’s t-test. * p < 0.05 vs. vehicle.
9. Conclusion
A novel AngII induced lncRNA Alivec regulates genes associated with chondrogenic transformation of VSMCs implicated in vascular dysfunction, which could lead to the identification of non-coding RNA based biomarkers and therapeutic targets for CVDs.
This entry is adapted from the peer-reviewed paper 10.3390/cells10102696
References
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237.
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164.
- Mulvihill, E.R.; Jaeger, J.; Sengupta, R.; Ruzzo, W.L.; Reimer, C.; Lukito, S.; Schwartz, S.M. Atherosclerotic Plaque Smooth Muscle Cells Have a Distinct Phenotype. Arter. Thromb. Vasc. Biol. 2004, 24, 1283–1289.
- Sahar, S.; Dwarakanath, R.S.; Reddy, M.A.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II enhances interleukin-18 me-diated inflammatory gene expression in vascular smooth muscle cells: A novel cross-talk in the pathogenesis of atherosclerosis. Circ. Res. 2005, 96, 1064–1071.
- Montezano, A.C.; Cat, A.N.D.; Rios, F.; Touyz, R.M. Angiotensin II and Vascular Injury. Curr. Hypertens. Rep. 2014, 16, 1–11.
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116.
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059.
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of Advanced Atherosclerotic Lesions in the Innominate Arteries of ApoE-Deficient Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1420–1425.
- Fakhry, M.; Roszkowska, M.; Briolay, A.; Bougault, C.; Guignandon, A.; Diaz-Hernandez, J.I.; Diaz-Hernandez, M.; Pikuła, S.; Buchet, R.; Hamade, E.; et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 643–653.
- Augstein, A.; Mierke, J.B.; Poitz, D.M.; Strasser, R.H. Sox9 is increased in arterial plaque and stenosis, associated with synthetic phenotype of vascular smooth muscle cells and causes alterations in extracellular matrix and calcification. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2526–2537.
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565.
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166.
- Ghanbari, M.; Peters, M.J.; de Vries, P.; Boer, C.; Van Rooij, J.G.J.; Lee, Y.-C.; Kumar, V.; Uitterlinden, A.G.; Ikram, M.A.; Wijmenga, C.; et al. A systematic analysis highlights multiple long non-coding RNAs associated with cardiometabolic disorders. J. Hum. Genet. 2018, 63, 431–446.
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2013, 15, 7–21.
- Simion, V.; Haemmig, S.; Feinberg, M.W. LncRNAs in vascular biology and disease. Vasc. Pharmacol. 2018, 114, 145–156.
- Leung, A.; Amaram, V.; Natarajan, R. Linking diabetic vascular complications with LncRNAs. Vasc. Pharmacol. 2019, 114, 139–144.
- Leung, A.; Trac, C.; Jin, W.; Lanting, L.; Akbany, A.; Sætrom, P.; Schones, D.E.; Natarajan, R. Novel Long Noncoding RNAs Are Regulated by Angiotensin II in Vascular Smooth Muscle Cells. Circ. Res. 2013, 113, 266–278.
- Tanwar, V.S.; Reddy, M.A.; Natarajan, R. Emerging Role of Long Non-Coding RNAs in Diabetic Vascular Complications. Front. Endocrinol. 2021, 12.
- Ballantyne, M.D.; Pinel, K.; Dakin, R.S.; Vesey, A.; A Diver, L.; MacKenzie, R.M.; Garcia, R.; Welsh, P.; Sattar, N.A.; Hamilton, G.; et al. Smooth Muscle Enriched Long Noncoding RNA (SMILR) Regulates Cell Proliferation. Circulation 2016, 133, 2050–2065.
- Bell, R.D.; Long, X.; Lin, M.; Bergmann, J.H.; Nanda, V.; Cowan, S.L.; Zhou, Q.; Han, Y.; Spector, D.L.; Zheng, D.; et al. Identification and Initial Functional Characterization of a Human Vascular Cell–Enriched Long Noncoding RNA. Arter. Thromb. Vasc. Biol. 2014, 34, 1249–1259.
- Das, S.; Senapati, P.; Chen, Z.; Reddy, M.A.; Ganguly, R.; Lanting, L.; Mandi, V.; Bansal, A.; Leung, A.; Zhang, S.; et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017, 8, 1–19.
- Shen, J.; Yang, M.; Jiang, H.; Ju, D.; Zheng, J.-P.; Xu, Z.; Liao, T.-D.; Li, L. Arterial injury promotes medial chondrogenesis in Sm22 knockout mice. Cardiovasc. Res. 2010, 90, 28–37.
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 1–16.
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738.
- Li, G.; Zheng, B.; Meszaros, L.B.; Vella, J.B.; Usas, A.; Matsumoto, T.; Huard, J. Identification and characterization of chondrogenic progenitor cells in the fascia of postnatal skeletal muscle. J. Mol. Cell Biol. 2011, 3, 369–377.
- Akiyama, H.; Kim, J.-E.; Nakashima, K.; Balmes, G.; Iwai, N.; Deng, J.M.; Zhang, Z.; Martin, J.F.; Behringer, R.R.; Nakamura, T.; et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 2005, 102, 14665–14670.
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110.
- Ferrè, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein–lncRNA interaction. Briefings Bioinform. 2015, 17, 106–116.
- Choi, H.-S.; Yim, S.-H.; Xu, H.-D.; Jung, S.-H.; Shin, S.-H.; Hu, H.-J.; Jung, C.-K.; Choi, J.Y.; Chung, Y.-J. Tropomyosin3 overexpression and a potential link to epithelial-mesenchymal transition in human hepatocellular carcinoma. BMC Cancer 2010, 10, 122.
- Kashiwada, K.; Nishida, W.; Hayashi, K.; Ozawa, K.; Yamanaka, Y.; Saga, H.; Yamashita, T.; Tohyama, M.; Shimada, S.; Sato, K.; et al. Coordinate Expression of α-Tropomyosin and Caldesmon Isoforms in Association with Phenotypic Modulation of Smooth Muscle Cells. J. Biol. Chem. 1997, 272, 15396–15404.
- Sobue, K.; Hayashi, K.; Nishida, W. Expressional regulation of smooth muscle cell-specific genes in association with phe-notypic modulation. Mol. Cell Biochem. 1999, 190, 105–118.
This entry is offline, you can click here to edit this entry!