c-Myc and the other protein family members (i.e., N-Myc and L-Myc), collectively known as “Myc”, are ubiquitous basic helix–loop–helix–leucine zipper (bHLH-LZ) transcription factors that are critical for several cellular processes during cancer genesis and progression. The importance of kinases in Myc regulation goes beyond their ability to phosphorylate the protein. Some kinases can also indirectly affect Myc protein stability by inducing the degradation of the ubiquitin ligase (PLK1 and PKA). Additionally, some kinases physically interact with Myc, protecting it from proteasomal degradation, such as Aurora-A in neuroblastoma.
- Myc
- kinases
- PLK1
- Aurora-A
- Aurora-B
- GSK-3
1. Introduction
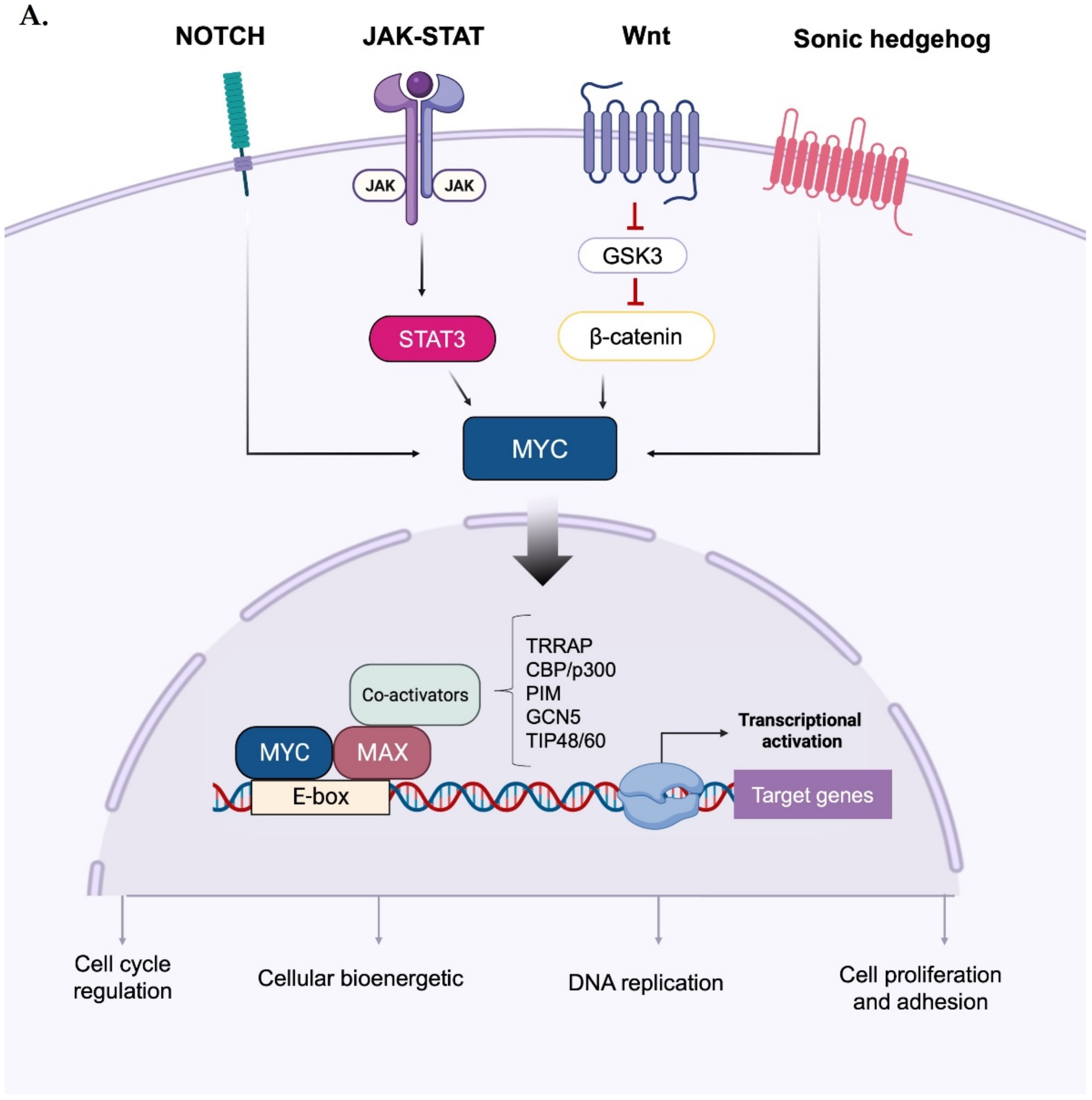
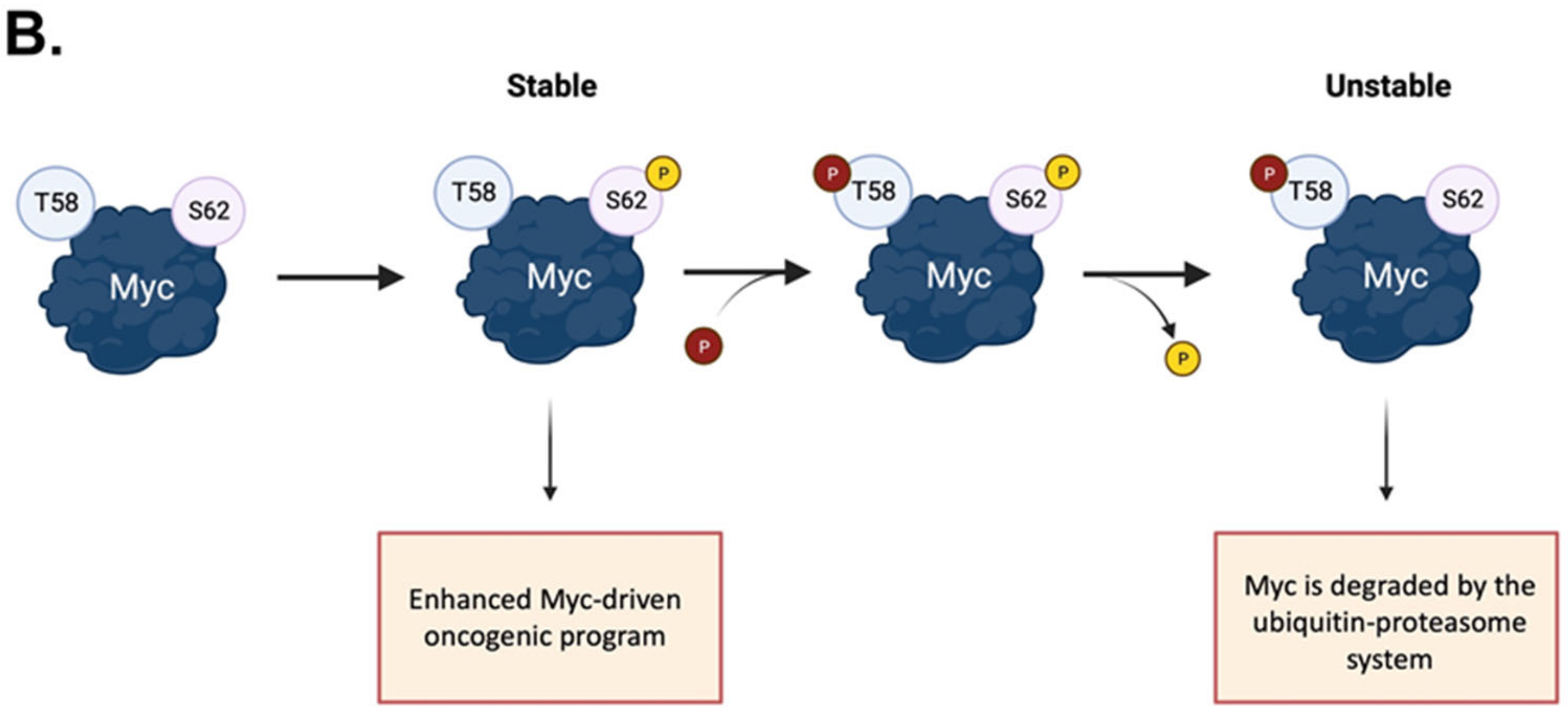
2. Direct and Indirect Myc Regulation by Mitotic Kinases
2.1. PLK1 Kinase
2.2. Aurora-A and Aurora-B Kinases
This entry is adapted from the peer-reviewed paper 10.3390/ijms24054746
References
- Scafuro, M.; Capasso, L.; Carafa, V.; Altucci, L.; Nebbioso, A. Gene transactivation and transrepression in myc-driven cancers. Int. J. Mol. Sci. 2021, 22, 3458.
- Sheiness, D.; Bishop, J.M. DNA and RNA from Uninfected Vertebrate Cells Contain Nucleotide Sequences Related to the Putative Transforming Gene of Avian Myelocytomatosis Virus. J. Virol. 1979, 31, 514–521.
- Roussel, M.; Saule, S.; Lagrou, C.; Rommens, C.; Beug, H.; Graf, T.; Stehelin, D. Three new types of viral oncogene of cellular origin specific for haematopoietic cell transformation. Nature 1979, 281, 452–455.
- Schwab, M.; Alitalo, K.; Klempnauer, K.-H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248.
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35 Pt 1, 359–367.
- Nau, M.M.; Brooks, B.J.; Battey, J.F.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.L.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73.
- Das, S.K.; Lewis, B.A.; Levens, D. MYC: A complex problem. Trends Cell Biol. 2023, 33, 235–246. Available online: http://www.ncbi.nlm.nih.gov/pubmed/35963793 (accessed on 22 February 2023).
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat Rev Cancer 2021, 21, 579–591.
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357.
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell. Biol. 2005, 6, 635–645.
- Hann, S.R. MYC cofactors: Molecular switches controlling diverse biological outcomes. Cold Spring Harb. Perspect. Med. 2014, 4, a014399.
- Hartl, M. The quest for targets executing MYC-dependent cell transformation. Front. Oncol. 2016, 6, 132.
- Tu, W.B.; Helander, S.; Pilstål, R.; Hickman, K.A.; Lourenco, C.; Jurisica, I.; Raught, B.; Wallner, B.; Sunnerhagen, M.; Penn, L.Z. Myc and its interactors take shape. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 469–483.
- Santinon, G.; Enzo, E.; Dupont, S. The sweet side of YAP/TAZ. Cell Cycle 2015, 14, 2543–2544.
- Rennoll, S. Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290.
- Zhang, S.; Li, Y.; Wu, Y.; Shi, K.; Bing, L.; Hao, J. Wnt/β-Catenin Signaling Pathway Upregulates c-Myc Expression to Promote Cell Proliferation of P19 Teratocarcinoma Cells. Anat. Rec. 2012, 295, 2104–2113.
- Liu, N.; Wang, S.; Cui, Y.; Shen, L.; Du, Y.; Li, G.; Zhang, B.; Wang, R. Sonic hedgehog elevates N-myc gene expression in neural stem cells. Neural Regen. Res. 2012, 7, 1703–1708.
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266.
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133.
- Huang, L.; Liu, D.; Wang, N.; Ling, S.; Tang, Y.; Wu, J.; Hao, L.; Luo, H.; Hu, X.; Sheng, L.; et al. Integrated genomic analysis identifies deregulated JAK/STAT-MYC-biosynthesis axis in aggressive NK-cell leukemia. Cell Res. 2018, 28, 172–186.
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells 2020, 9, 217.
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039.
- Battey, J.; Moulding, C.; Taub, R.; Murphy, W.; Stewart, T.; Potter, H.; Lenoir, G.; Leder, P. The human c-myc oncogene: Structural consequences of translocation into the igh locus in Burkitt lymphoma. Cell 1983, 34, 779–787.
- Liu, J.; Levens, D. Making Myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32.
- Dani, C.; Blanchard, J.M.; Piechaczyk, M.; El Sabouty, S.; Marty, L.; Jeanteur, P. Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. USA 1984, 81, 7046–7050.
- Hann, S.R.; Eisenman, R.N. Proteins encoded by the human c-myc oncogene: Differential expression in neoplastic cells. Mol. Cell. Biol. 1984, 4, 2486–2497.
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, 1–16.
- González-Prieto, R.; Cuijpers, S.A.G.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C.O. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872.
- Kalkat, M.; Chan, P.-K.; Wasylishen, A.R.; Srikumar, T.; Kim, S.S.; Ponzielli, R.; Bazett-Jones, D.P.; Raught, B.; Penn, L.Z. Identification of c-MYC SUMOylation by mass spectrometry. PLoS ONE 2014, 9, e115337.
- Sabò, A.; Doni, M.; Amati, B. SUMOylation of Myc-family proteins. PLoS ONE 2014, 9, e91072.
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Clin. Cancer Res. 2016, 22, 5539–5552.
- Höllein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090.
- Suna, X.X.; Chena, Y.; Sua, Y.; Wanga, X.; Chauhana, K.M.; Lianga, J.; Daniel, C.J.; Sears, R.C.; Dai, M.-S. SUMO protease SENP1 deSUMOylates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2018, 115, 10983–10988.
- Sun, X.X.; Li, Y.; Sears, R.C.; Dai, M.S. Targeting the MYC Ubiquitination-Proteasome Degradation Pathway for Cancer Therapy. Front. Oncol. 2021, 11, 679445.
- Henriksson, M.; Bakardjiev, A.; Klein, G.; Luscher, B. Phosphorylation sites mapping in the N-terminal domain of c-myc modulate its transforming potential. Oncogene 1993, 8, 3199–3209.
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006, 16, 288–302.
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519.
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J. Biol. Chem. 2003, 278, 51606–51612.
- Bahram, F.; Von Der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110.
- Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Destruction of Myc by ubiquitin-mediated proteolysis: Cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999, 18, 717–726.
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936.
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811.
- Johnson, N.A. Functional and clinical impact of MYC mutations in diffuse large B cell lymphomas. Transl Cancer Res. 2016, 5, S257–S260.
- Huang, Z.; Traugh, J.A.; Bishop, J.M. Negative Control of the Myc Protein by the Stress-Responsive Kinase Pak2. Mol. Cell. Biol. 2004, 24, 1582–1594.
- Macek, P.; Cliff, M.J.; Embrey, K.J.; Holdgate, G.A.; Nissink, J.W.M.; Panova, S.; Waltho, J.P.; Davies, R.A. Myc phosphorylation in its basic helix?loop?helix region destabilizes transient-helical structures, disrupting Max and DNA binding. J. Biol. Chem. 2018, 293, 9301–9310.
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 2010, 107, 163–224.
- Herbst, A.; Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Multiple cell-type-specific elements regulate Myc protein stability. Oncogene 2004, 23, 3863–3871.
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A Conserved Myc Protein Domain, MBIV, Regulates DNA Binding, Apoptosis, Transformation, and G 2 Arrest. Mol. Cell. Biol. 2006, 26, 4226–4239.
- Vervoorts, J.; Lüscher-Firzlaff, J.; Lüscher, B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006, 281, 34725–34729.
- Xiao, D.; Yue, M.; Su, H.; Ren, P.; Jiang, J.; Li, F.; Hu, Y.; Du, H.; Liu, H.; Qing, G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol. Cell 2016, 64, 493–506.
- Popov, N.; Schülein, C.; Jaenicke, L.A.; Eilers, M. Ubiquitylation of the amino terminus of Myc by SCFβ-TrCP antagonizes SCFFbw7-mediated turnover. Nat. Cell Biol. 2010, 12, 973–981.
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78.
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; et al. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis. Cancer Cell 2020, 37, 200–215.e5.
- Naso, F.D.; Boi, D.; Ascanelli, C.; Pamfil, G.; Lindon, C.; Paiardini, A.; Guarguaglini, G. Nuclear localisation of Aurora-A: Its regulation and significance for Aurora-A functions in cancer. Oncogene 2021, 40, 3917–3928.
- Chan, G.K.L.; Maisel, S.; Hwang, Y.C.; Wolber, R.R.B.; Vu, P.; Patra, C.; Bouhaddou, M.; Kenerson, H.L.; Yeung, R.S.; Swaney, D.L.; et al. Oncogenic PKA signaling stabilizes MYC oncoproteins via an aurora kinase A-dependent mechanism. bioRxiv 2021.
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819.
- Zippo, A.; De Robertis, A.; Serafini, R.; Oliviero, S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 2007, 9, 932–944.
- Devaiah, B.N.; Mu, J.; Akman, B.; Uppal, S.; Weissman, J.D.; Cheng, D.; Baranello, L.; Nie, Z.; Levens, D.; Singer, D.S. MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. USA 2020, 117, 13457–13467.
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494.
- Combes, G.; Alharbi, I.; Braga, L.G.; Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 2017, 36, 4819–4827.
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016.
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747.
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32.
- Montaudon, E.; Nikitorowicz-Buniak, J.; Sourd, L.; Morisset, L.; EL Botty, R.; Huguet, L.; Dahmani, A.; Painsec, P.; Nemati, F.; Vacher, S.; et al. PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance. Nat. Commun. 2020, 11, 4053.
- Nieto-Jimenez, C.; Galan-Moya, E.M.; Corrales-Sanchez, V.; Noblejas-Lopez, M.D.M.; Burgos, M.; Domingo, B.; Montero, J.C.; Gomez-Juarez, M.; Picazo-Martinez, M.G.; Esparis-Ogando, A.; et al. Inhibition of the mitotic kinase PLK1 overcomes therapeutic resistance to BET inhibitors in triple negative breast cancer. Cancer Lett. 2020, 491, 50–59.
- Zhang, Z.; Cheng, L.; Li, J.; Qiao, Q.; Karki, A.; Allison, D.B.; Shaker, N.; Li, K.; Utturkar, S.M.; Atallah Lanman, N.M.; et al. Targeting Plk1 sensitizes pancreatic cancer to immune checkpoint therapy. Cancer Res. 2022, 82, 3532–3548.
- Bibi, N.; Parveen, Z.; Rashid, S. Identification of Potential Plk1 Targets in a Cell-Cycle Specific Proteome through Structural Dynamics of Kinase and Polo Box-Mediated Interactions. PLoS ONE 2013, 8, e70843.
- Liu, J.; Zhang, C. The equilibrium of ubiquitination and deubiquitination at PLK1 regulates sister chromatid separation. Cell. Mol. Life Sci. 2017, 74, 2127–2134.
- Raab, M.; Matthess, Y.; Raab, C.A.; Gutfreund, N.; Dötsch, V.; Becker, S.; Sanhaji, M.; Strebhardt, K. A dimerization-dependent mechanism regulates enzymatic activation and nuclear entry of PLK1. Oncogene 2021, 41, 372–386.
- Xu, J.; Shen, C.; Wang, T.; Quan, J. Structural basis for the inhibition of Polo-like kinase 1. Nat Struct Mol Biol. 2013, 20, 1047–1053.
- Beck, J.; Maerki, S.; Posch, M.; Metzger, T.; Persaud, A.; Scheel, H.; Hofmann, K.; Rotin, D.; Pedrioli, P.; Swedlow, J.R.; et al. Ubiquitylation-dependent localization of PLK1 in mitosis. Nat. Cell Biol. 2013, 15, 430–439.
- Kachaner, D.; Garrido, D.; Mehsen, H.; Normandin, K.; Lavoie, H.; Archambault, V. Coupling of Polo kinase activation to nuclear localization by a bifunctional NLS is required during mitotic entry. Nat. Commun. 2017, 8, 1701.
- Zhou, J.; Yang, Q.; Lu, L.; Tuo, Z.; Shou, Z.; Cheng, J. Plk1 inhibition induces immunogenic cell death and enhances immunity against nsclc. Int. J. Med. Sci. 2021, 18, 3516–3525.
- Fu, Z.; Wen, D. The emerging role of polo-like kinase 1 in epithelial-mesenchymal transition and tumor metastasis. Cancers 2017, 9, 131.
- Song, R.; Hou, G.; Yang, J.; Yuan, J.; Wang, C.; Chai, T.; Liu, Z. Effects of PLK1 on proliferation, invasion and metastasis of gastric cancer cells through epithelial-mesenchymal transition. Oncol. Lett. 2018, 16, 5739–5744.
- Gao, Z.; Man, X.; Li, Z.; Bi, J.; Liu, X.; Li, Z.; Li, J.; Zhang, Z.; Kong, C. PLK1 promotes proliferation and suppresses apoptosis of renal cell carcinoma cells by phosphorylating MCM3. Cancer Gene Ther. 2020, 27, 412–423.
- Luo, P.; Yan, H.; Du, J.; Chen, X.; Shao, J.; Zhang, Y.; Xu, Z.; Jin, Y.; Lin, N.; Yang, B.; et al. PLK1 (polo like kinase 1)-dependent autophagy facilitates gefitinib-induced hepatotoxicity by degrading COX6A1 (cytochrome c oxidase subunit 6A1). Autophagy 2021, 17, 3221–3237.
- Oon, M.L.; Hoppe, M.M.; Fan, S.; Phyu, T.; Phuong, H.M.; Tan, S.-Y.; Hue, S.S.-S.; Wang, S.; Poon, L.M.; Chan, H.L.E.; et al. The contribution of MYC and PLK1 expression to proliferative capacity in diffuse large B-cell lymphoma. Leuk Lymphoma 2019, 60, 3214–3224.
- Ren, Y.; Bi, C.; Zhao, X.; Lwin, T.; Wang, C.; Yuan, J.; Silva, A.S.; Shah, B.D.; Fang, B.; Li, T.; et al. PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J. Clin. Investig. 2018, 128, 5531–5548.
- Yu, Z.; Deng, P.; Chen, Y.; Liu, S.; Chen, J.; Yang, Z.; Chen, J.; Fan, X.; Wang, P.; Cai, Z.; et al. Inhibition of the PLK1-Coupled Cell Cycle Machinery Overcomes Resistance to Oxaliplatin in Colorectal Cancer. Adv. Sci. 2021, 8, 2100759.
- Murga-Zamalloa, C.; Polk, A.; Hanel, W.; Chowdhury, P.; Brown, N.; Hristov, A.C.; Bailey, N.G.; Wang, T.; Phillips, T.; Devata, S.; et al. Polo-like-kinase 1 (PLK-1) and c-myc inhibition with the dual kinase-bromodomain inhibitor volasertib in aggressive lymphomas. Oncotarget 2017, 8, 114474–114480.
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.J.; Wee, Z.N.; et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171.
- Padmanabhan, A.; Li, X.; Bieberich, C.J. Protein kinase a regulates MYC protein through transcriptional and post-translational mechanisms in a catalytic subunit isoform-specific manner. J. Biol. Chem. 2013, 288, 14158–14169.
- Wang, D.; Pierce, A.; Veo, B.; Fosmire, S.; Danis, E.; Donson, A.; Venkataraman, S.; Vibhakar, R. A regulatory loop of FBXW7-MYC-PLK1 controls tumorigenesis of MYC-driven medulloblastoma. Cancers 2021, 13, 387.
- Mo, H.; He, J.; Yuan, Z.; Wu, Z.; Liu, B.; Lin, X.; Guan, J. PLK1 contributes to autophagy by regulating MYC stabilization in osteosarcoma cells. Onco Targets Ther. 2019, 12, 7527–7536.
- Quartuccio, S.M.; Schindler, K. Functions of Aurora kinase C in meiosis and cancer. Front. Cell Dev. Biol. 2015, 3, 50.
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7.
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195.
- Gallini, S.; Carminati, M.; De Mattia, F.; Pirovano, L.; Martini, E.; Oldani, A.; Asteriti, I.A.; Guarguaglini, G.; Mapelli, M. NuMA phosphorylation by aurora-a orchestrates spindle orientation. Curr. Biol. 2016, 26, 458–469.
- Polverino, F.; Naso, F.D.; Asteriti, I.A.; Palmerini, V.; Singh, D.; Valente, D.; Bird, A.W.; Rosa, A.; Mapelli, M.; Guarguaglini, G. The Aurora-A/TPX2 Axis Directs Spindle Orientation in Adherent Human Cells by Regulating NuMA and Microtubule Stability. Curr. Biol. 2021, 31, 658–667.e5.
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell. Biol. 2012, 13, 789–803.
- Van Der Horst, A.; Lens, S.M.A. Cell division: Control of the chromosomal passenger complex in time and space. Chromosoma 2014, 123, 25–42.
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954.
- Gautschi, O.; Heighway, J.; Mack, P.C.; Purnell, P.R.; Lara, P.N.; Gandara, D.R. Aurora kinases as anticancer drug targets. Clin. Cancer Res. 2008, 14, 1639–1648.
- Lens, S.M.A.; Voest, E.E.; Medema, R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841.
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847.
- Asteriti, I.A.; Rensen, W.M.; Lindon, C.; Lavia, P.; Guarguaglini, G. The Aurora-A/TPX2 complex: A novel oncogenic holoenzyme? Biochim Biophys Act-Rev. Cancer 2010, 1806, 230–239.
- Wan, X.-B.; Long, Z.-J.; Yan, M.; Xu, J.; Xia, L.-P.; Liu, L.; Zhao, Y.; Huang, X.-F.; Wang, X.-R.; Zhu, X.-F.; et al. Inhibition of Aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis 2008, 29, 1930–1937.
- Liu, X.; Li, Z.; Song, Y.; Wang, R.; Han, L.; Wang, Q.; Jiang, K.; Kang, C.; Zhang, Q. AURKA induces EMT by regulating histone modification through Wnt/ß-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget 2016, 7, 33152–33164.
- Xia, Z.; Wei, P.; Zhang, H.; Ding, Z.; Yang, L.; Huang, Z.; Zhang, N. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of β-catenin/Wnt Signaling. Mol Cancer Res. 2013, 11, 1101–1111.
- Lin, Z.-Z.; Jeng, Y.-M.; Hu, F.-C.; Pan, H.-W.; Tsao, H.-W.; Lai, P.-L.; Lee, P.-H.; Cheng, A.-L.; Hsu, H.-C. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer 2010, 10, 461.
- Vischioni, B.; Oudejans, J.J.; Vos, W.; Rodriguez, J.A.; Giaccone, G. Frequent overexpression of aurora B kinase, a novel drug target, in non-small cell lung carcinoma patients. Mol. Cancer Ther. 2006, 5, 2905–2913.
- Qi, G.; Ogawa, I.; Kudo, Y.; Miyauchi, M.; Siriwardena, B.S.M.S.; Shimamoto, F.; Tatsuka, M.; Takata, T. Aurora-B expression and its correlation with cell proliferation and metastasis in oral cancer. Virchows Arch. 2007, 450, 297–302.
- Den Hollander, J.; Rimpi, S.; Doherty, J.R.; Rudelius, M.; Buck, A.; Hoellein, A.; Kremer, M.; Graf, N.; Scheerer, M.; Hall, M.A.; et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010, 116, 1498–1505.
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.-W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753.
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495.
- Vader, G.; Lens, S.M.A. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta-Rev. Cancer 2008, 1786, 60–72.
- Büchel, G.; Carstensen, A.; Mak, K.-Y.; Roeschert, I.; Leen, E.; Sumara, O.; Hofstetter, J.; Herold, S.; Kalb, J.; Baluapuri, A.; et al. Association with Aurora-A Controls N-MYC-Dependent Promoter Escape and Pause Release of RNA Polymerase II during the Cell Cycle. Cell Rep. 2017, 21, 3483–3497.
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731.
- Lu, L.; Han, H.; Tian, Y.; Li, W.; Zhang, J.; Feng, M.; Li, Y. Aurora kinase A mediates c-Myc’s oncogenic effects in hepatocellular carcinoma. Mol. Carcinog. 2015, 54, 1467–1479.
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180.
- Hsueh, K.W.; Fu, S.L.; Huang, C.Y.F.; Lin, C.H. Aurora-A phosphorylates hnRNPK and disrupts its interaction with p53. FEBS Lett. 2011, 585, 2671–2675.
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. p53-Dependent Transcriptional Repression of c-myc Is Required for G1 Cell Cycle Arrest. Mol. Cell. Biol. 2005, 25, 7423–7431.
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624–638.e8.
- Bayliss, R.; Sardon, T.; Ebert, J.; Lindner, D.; Vernos, I.; Conti, E. Determinants for Aurora-A activation and Aurora-B discrimination by TPX2. Cell Cycle 2004, 3, 402–405.
- Fu, J.; Bian, M.; Liu, J.; Jiang, Q.; Zhang, C. A single amino acid change converts Aurora-A into Aurora-B-like kinase in terms of partner specificity and cellular function. Proc. Natl. Acad. Sci. USA 2009, 106, 6939–6944.
- Eyers, P.A.; Churchill, M.E.A.; Maller, J.L. The Aurora A and Aurora B protein kinases: A single amino acid difference controls intrinsic activity and activation by TPX2. Cell Cycle 2005, 4, 784–789.
- Hans, F.; Skoufias, D.A.; Dimitrov, S.; Margolis, R.L. Molecular distinctions between Aurora A and B: A single residue change transforms Aurora A into correctly localized and functional Aurora B. Mol. Biol. Cell 2009, 20, 3491–3502.
- DeLuca, K.F.; Meppelink, A.; Broad, A.J.; Mick, J.E.; Peersen, O.B.; Pektas, S.; Lens, S.M.; DeLuca, J.G. Aurora A kinase phosphorylates Hec1 to regulate metaphase kinetochore-microtubule dynamics. J. Cell Biol. 2018, 217, 163–177.
- Berwanger, B.; Hartmann, O.; Bergmann, E.; Bernard, S.; Nielsen, D.; Krause, M.; Kartal, A.; Flynn, D.; Wiedemeyer, R.; Schwab, M.; et al. Loss of a FYN-regulated differentiation and growth arrest pathway in advanced stage neuroblastoma. Cancer Cell 2002, 2, 377–386.
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89.
- Bogen, D.; Wei, J.S.; Azorsa, D.O.; Ormanoglu, P.; Buehler, E.; Guha, R.; Keller, J.M.; Griner, L.A.M.; Ferrer, M.; Song, Y.K.; et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget 2015, 6, 35247–35262.
- Borah, N.A.; Sradhanjali, S.; Barik, M.R.; Jha, A.; Tripathy, D.; Kaliki, S.; Rath, S.; Raghav, S.K.; Patnaik, S.; Mittal, R.; et al. Aurora kinase B expression, its regulation and therapeutic targeting in human retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 16.
- Li, M.; Sun, C.; Bu, X.; Que, Y.; Zhang, L.; Zhang, Y.; Zhang, L.; Lu, S.; Huang, J.; Zhu, J.; et al. ISL1 promoted tumorigenesis and EMT via Aurora kinase A-induced activation of PI3K/AKT signaling pathway in neuroblastoma. Cell Death Dis. 2021, 12, 620.
- Dar, A.A.; Belkhiri, A.; El-Rifai, W. The aurora kinase A regulates GSK-3β in gastric cancer cells. Oncogene 2009, 28, 866–875.
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.-M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62.