The liver is one of the main organs that contributes to the regulation of blood glucose levels by releasing the glucose into circulation when blood glucose levels are low and stores along skeletal muscles excess of glucose as glycogen in postprandial states. In mammals, the liver is the central organ for fatty acid metabolism and a key player in glucose metabolism. The liver acts as a crossroad, which metabolically connects various organs, especially skeletal muscle, and adipose tissues, in the endeavor to maintain the long-term energy supply to the body. The regulation of liver glucose metabolism and lipid metabolism is tightly controlled by dietary, hormonal, and neural signals through the activation of various transcriptional factors and coactivators.
- fatty liver
- obesity
- type 2 diabetes
1. Introduction
2. Regulation of Liver Metabolism by Transcription Factors
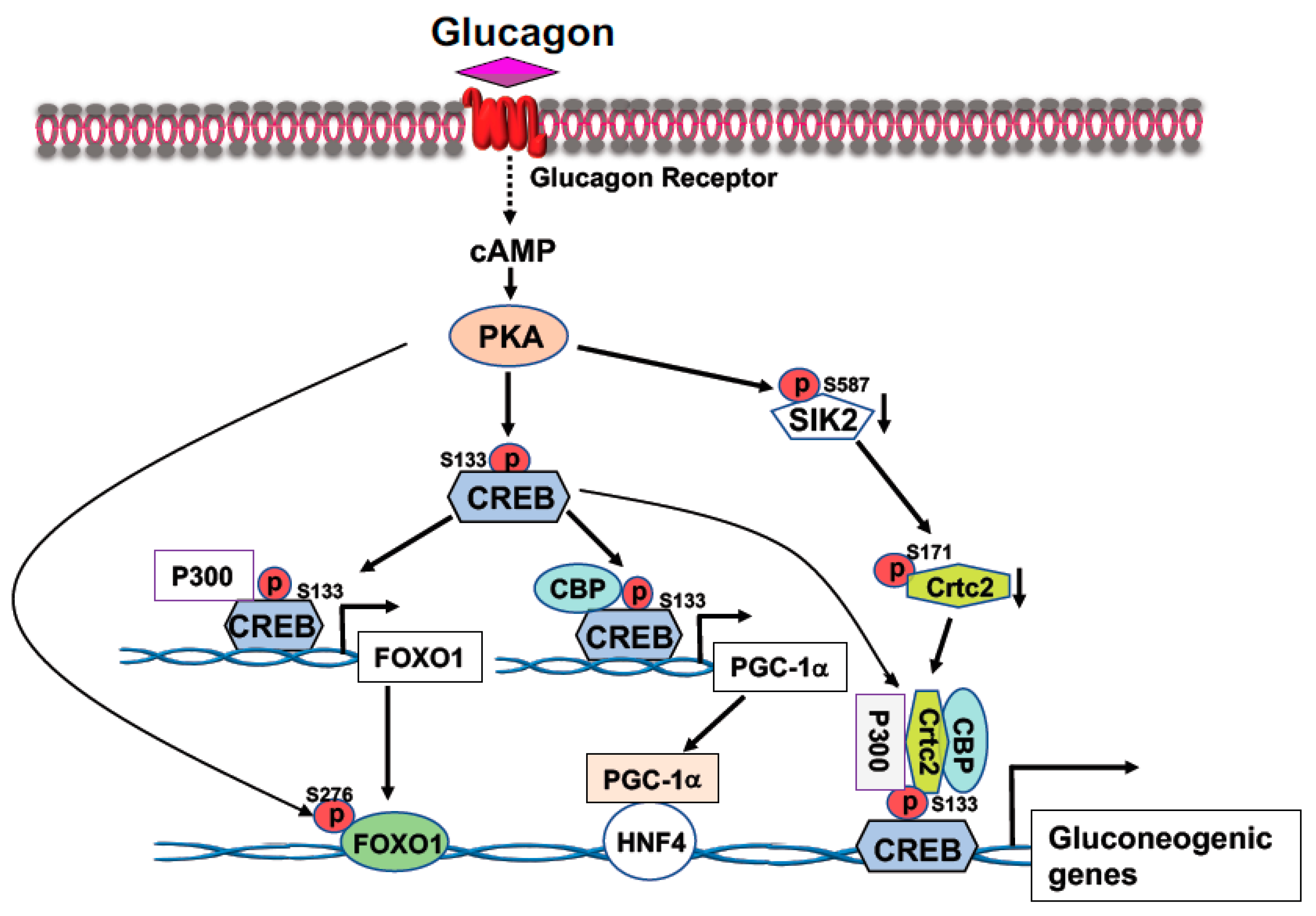
2.1. Cyclic AMP Response-Element Binding Protein (CREB)
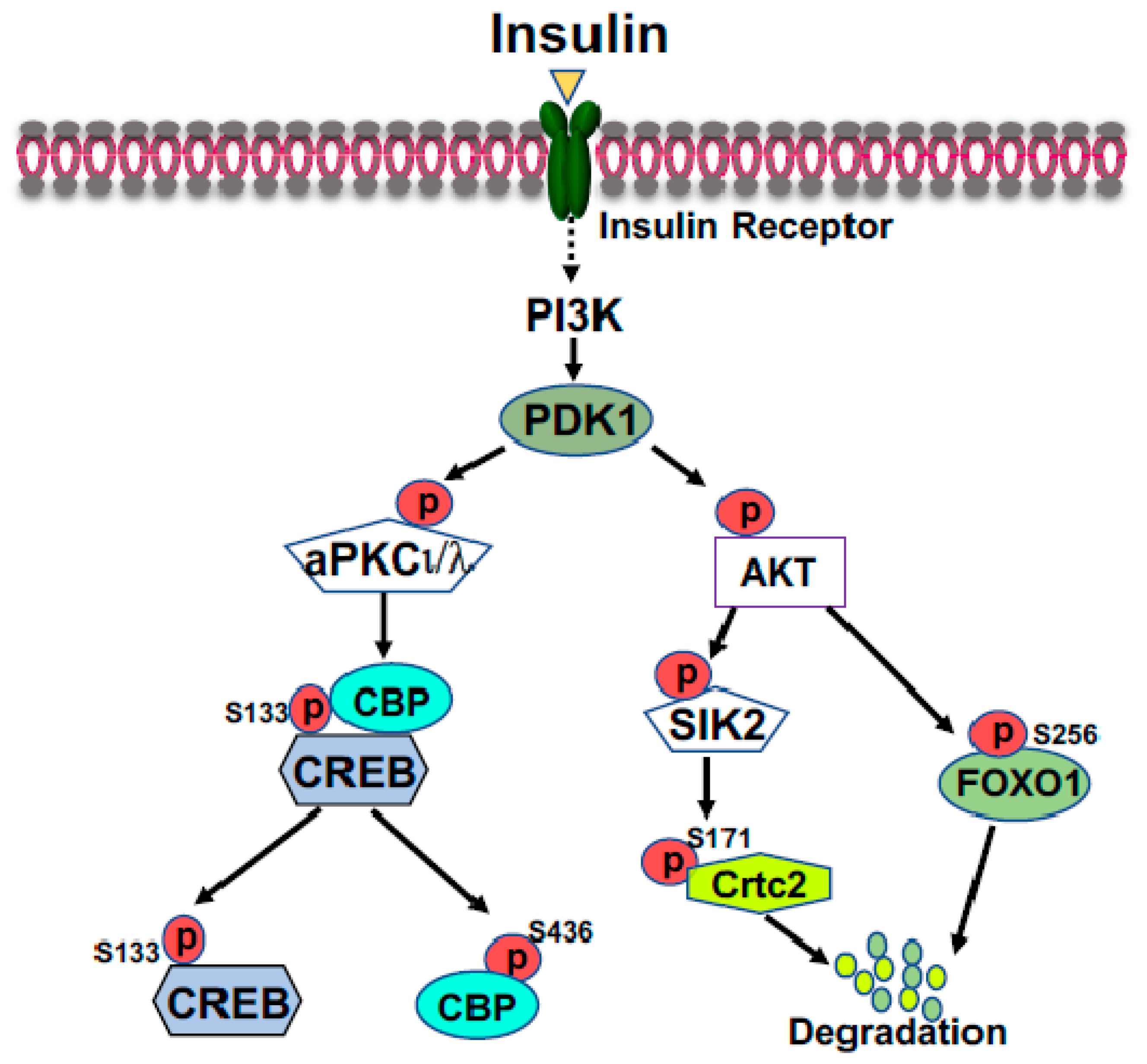
2.2. FOXO1
2.3. Carbohydrate Response Element Binding Protein (ChREBP)
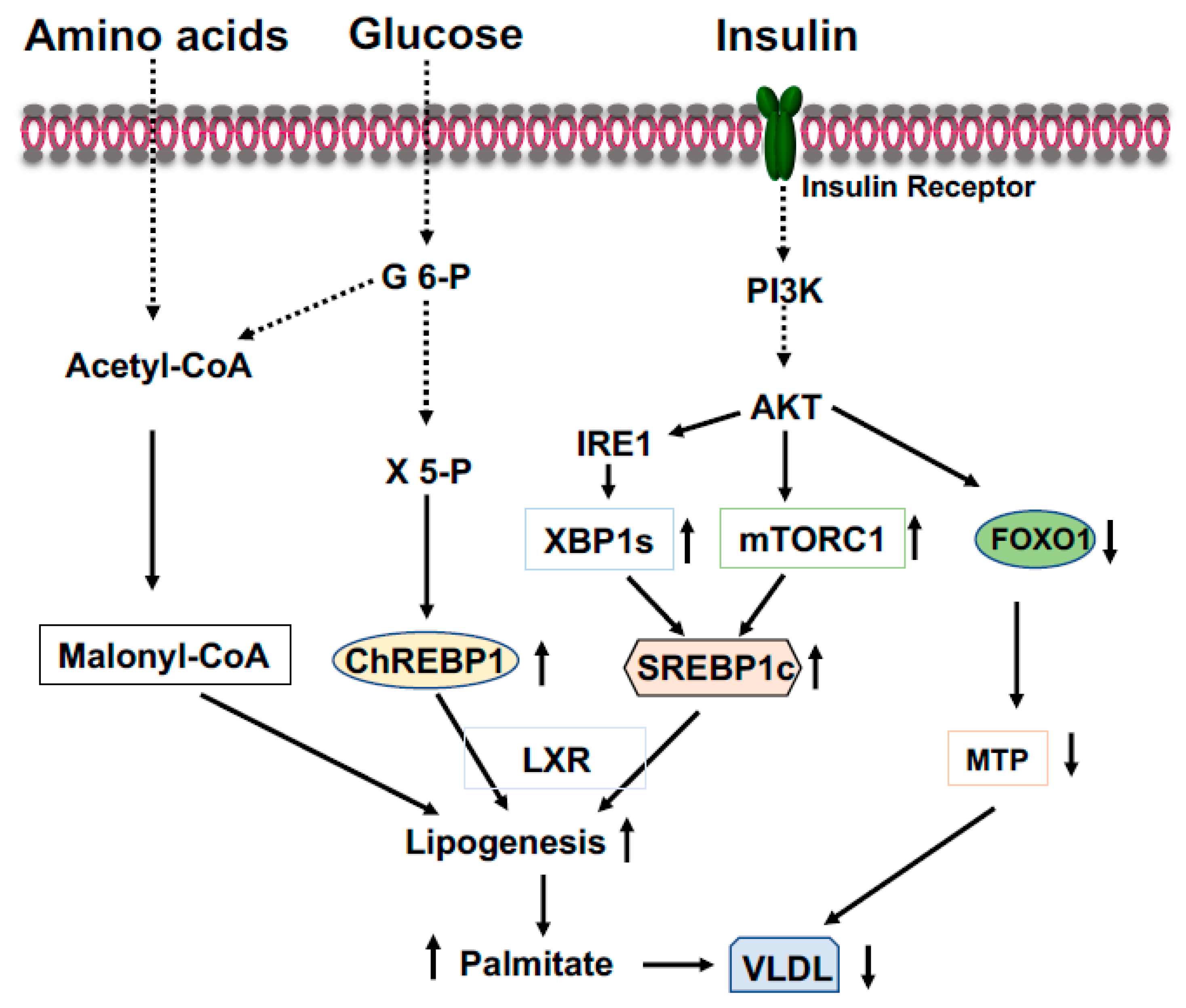
2.4. Sterol Regulatory Element-Binding Protein-1 (SREBP-1)
2.5. X-Box Binding Protein 1 (XBP1)
2.6. Nuclear Receptors
2.6.1. Peroxisome Proliferator Activated Receptors (PPARs)
2.6.2. Liver X Receptors (LXR)
2.6.3. Farnesoid X Receptors
2.6.4. Hepatocyte Nuclear Factor 4 (HNF4)
3. Regulation of Liver Metabolism by Coactivators and Corepressors
3.1. Regulation of Liver Metabolism of Coactivators CBP and P300
3.2. Regulation of Liver Metabolism of CREB-Regulated Transcription Coactivator 2 (CRTC2)
3.3. Regulation of Liver Metabolism of PGC-1α
3.4. Regulation of Liver Metabolism of Corepressors
This entry is adapted from the peer-reviewed paper 10.3390/life13020515
References
- Doria, A.; Patti, M.E.; Kahn, C.R. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008, 8, 186–200.
- Masuoka, H.C.; Chalasani, N. Nonalcoholic fatty liver disease: An emerging threat to obese and diabetic individuals. Ann. N. Y. Acad. Sci. 2013, 1281, 106–122.
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Despres, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010.
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790.
- Dabelea, D.; Mayer-Davis, E.J.; Saydah, S.; Imperatore, G.; Linder, B.; Divers, J.; Bell, R.; Badaru, A.; Talton, J.W.; Crume, T.; et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 2014, 311, 1778–1786.
- Papatheodorou, K.; Banach, M.; Bekiari, E.; Rizzo, M.; Edmonds, M. Complications of Diabetes 2017. J. Diabetes Res. 2018, 2018, 3086167.
- Khan, R.M.M.; Chua, Z.J.Y.; Tan, J.C.; Yang, Y.; Liao, Z.; Zhao, Y. From Pre-Diabetes to Diabetes: Diagnosis, Treatments and Translational Research. Medicina 2019, 55, 546.
- Zhao, Y.; Xing, H.; Wang, X.; Ou, W.; Zhao, H.; Li, B.; Li, Y.; Duan, Y.; Zhuang, L.; Li, W.; et al. Management of Diabetes Mellitus in Patients with Chronic Liver Diseases. J. Diabetes Res. 2019, 2019, 6430486.
- He, L. Alterations of Gut Microbiota by Overnutrition Impact Gluconeogenic Gene Expression and Insulin Signaling. Int. J. Mol. Sci. 2021, 22, 2121.
- Koh, G.C.; Peacock, S.J.; van der Poll, T.; Wiersinga, W.J. The impact of diabetes on the pathogenesis of sepsis. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 379–388.
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626.
- Vannucci, R.C.; Vannucci, S.J. Hypoglycemic brain injury. Semin. Neonatol. 2001, 6, 147–155.
- Desouza, C.; Salazar, H.; Cheong, B.; Murgo, J.; Fonseca, V. Association of hypoglycemia and cardiac ischemia: A study based on continuous monitoring. Diabetes Care 2003, 26, 1485–1489.
- Einarsdottir, A.B.; Stefansson, E. Prevention of diabetic retinopathy. Lancet 2009, 373, 1316–1318.
- Bellentani, S.; Marino, M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann. Hepatol. 2009, 8 (Suppl. S1), S4–S8.
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151.
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111.
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646.
- Muraoka, M.; Fukushima, A.; Viengchareun, S.; Lombes, M.; Kishi, F.; Miyauchi, A.; Kanematsu, M.; Doi, J.; Kajimura, J.; Nakai, R.; et al. Involvement of SIK2/TORC2 signaling cascade in the regulation of insulin-induced PGC-1alpha and UCP-1 gene expression in brown adipocytes. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1430–E1439.
- He, L.; Naik, K.; Meng, S.; Cao, J.; Sidhaye, A.R.; Ma, A.; Radovick, S.; Wondisford, F.E. Transcriptional co-activator p300 maintains basal hepatic gluconeogenesis. J. Biol. Chem. 2012, 287, 32069–32077.
- Erion, D.M.; Ignatova, I.D.; Yonemitsu, S.; Nagai, Y.; Chatterjee, P.; Weismann, D.; Hsiao, J.J.; Zhang, D.; Iwasaki, T.; Stark, R.; et al. Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metab. 2009, 10, 499–506.
- Peng, S.; Li, W.; Hou, N.; Huang, N. A Review of FoxO1-Regulated Metabolic Diseases and Related Drug Discoveries. Cells 2020, 9, 184.
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425.
- Furuyama, T.; Nakazawa, T.; Nakano, I.; Mori, N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 2000, 349, 629–634.
- Brent, M.M.; Anand, R.; Marmorstein, R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 2008, 16, 1407–1416.
- Lalmansingh, A.S.; Karmakar, S.; Jin, Y.; Nagaich, A.K. Multiple modes of chromatin remodeling by Forkhead box proteins. Biochim. Biophys. Acta 2012, 1819, 707–715.
- Zhang, X.; Gan, L.; Pan, H.; Guo, S.; He, X.; Olson, S.T.; Mesecar, A.; Adam, S.; Unterman, T.G. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 2002, 277, 45276–45284.
- Xie, Q.; Chen, J.; Yuan, Z. Post-translational regulation of FOXO. Acta Biochim. Biophys. Sin. 2012, 44, 897–901.
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; De Pinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76.
- Zhang, K.; Li, L.; Qi, Y.; Zhu, X.; Gan, B.; DePinho, R.A.; Averitt, T.; Guo, S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012, 153, 631–646.
- Wu, Y.; Pan, Q.; Yan, H.; Zhang, K.; Guo, X.; Xu, Z.; Yang, W.; Qi, Y.; Guo, C.A.; Hornsby, C.; et al. Novel Mechanism of Foxo1 Phosphorylation in Glucagon Signaling in Control of Glucose Homeostasis. Diabetes 2018, 67, 2167–2182.
- Guo, S.; Dunn, S.L.; White, M.F. The reciprocal stability of FOXO1 and IRS2 creates a regulatory circuit that controls insulin signaling. Mol. Endocrinol. 2006, 20, 3389–3399.
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J. Clin. Investig. 2008, 118, 2347–2364.
- Altomonte, J.; Cong, L.; Harbaran, S.; Richter, A.; Xu, J.; Meseck, M.; Dong, H.H. Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J. Clin. Investig. 2004, 114, 1493–1503.
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 474–485.
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286.
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715.
- Morigny, P.; Houssier, M.; Mairal, A.; Ghilain, C.; Mouisel, E.; Benhamed, F.; Masri, B.; Recazens, E.; Denechaud, P.D.; Tavernier, G.; et al. Interaction between hormone-sensitive lipase and ChREBP in fat cells controls insulin sensitivity. Nat. Metab. 2019, 1, 133–146.
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferre, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209.
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Glucose activates ChREBP by increasing its rate of nuclear entry and relieving repression of its transcriptional activity. J. Biol. Chem. 2008, 283, 24029–24038.
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386.
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730.
- Amemiya-Kudo, M.; Shimano, H.; Hasty, A.H.; Yahagi, N.; Yoshikawa, T.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J. Lipid Res. 2002, 43, 1220–1235.
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131.
- Shao, W.; Espenshade, P.J. Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). J. Biol. Chem. 2014, 289, 7547–7557.
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661.
- Matsumoto, E.; Ishihara, A.; Tamai, S.; Nemoto, A.; Iwase, K.; Hiwasa, T.; Shibata, S.; Takiguchi, M. Time of day and nutrients in feeding govern daily expression rhythms of the gene for sterol regulatory element-binding protein (SREBP)-1 in the mouse liver. J. Biol. Chem. 2010, 285, 33028–33036.
- Tian, J.; Goldstein, J.L.; Brown, M.S. Insulin induction of SREBP-1c in rodent liver requires LXRalpha-C/EBPbeta complex. Proc. Natl. Acad. Sci. USA 2016, 113, 8182–8187.
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500.
- Yabe, D.; Komuro, R.; Liang, G.; Goldstein, J.L.; Brown, M.S. Liver-specific mRNA for Insig-2 down-regulated by insulin: Implications for fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3155–3160.
- Shimano, H.; Horton, J.D.; Shimomura, I.; Hammer, R.E.; Brown, M.S.; Goldstein, J.L. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J. Clin. Investig. 1997, 99, 846–854.
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871.
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529.
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789.
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461.
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.S.; Klein, S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009, 58, 693–700.
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferre, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 2009, 119, 1201–1215.
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496.
- Sha, H.; He, Y.; Chen, H.; Wang, C.; Zenno, A.; Shi, H.; Yang, X.; Zhang, X.; Qi, L. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009, 9, 556–564.
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96.
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466.
- Ning, J.; Hong, T.; Ward, A.; Pi, J.; Liu, Z.; Liu, H.Y.; Cao, W. Constitutive role for IRE1alpha-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology 2011, 152, 2247–2255.
- Cao, J.; Peng, J.; An, H.; He, Q.; Boronina, T.; Guo, S.; White, M.F.; Cole, P.A.; He, L. Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat. Commun. 2017, 8, 131.
- Peng, J.; He, L. IRS posttranslational modifications in regulating insulin signaling. J. Mol. Endocrinol. 2018, 60, R1–R8.
- Peng, J.; Qin, C.; Ramatchandirin, B.; Pearah, A.; Guo, S.; Hussain, M.; Yu, L.; Wondisford, F.E.; He, L. Activation of the canonical ER stress IRE1-XBP1 pathway by insulin regulates glucose and lipid metabolism. J. Biol. Chem. 2022, 298, 102283.
- Dhiman, V.K.; Bolt, M.J.; White, K.P. Nuclear receptors in cancer—Uncovering new and evolving roles through genomic analysis. Nat. Rev. Genet. 2018, 19, 160–174.
- Rastinejad, F.; Huang, P.; Chandra, V.; Khorasanizadeh, S. Understanding nuclear receptor form and function using structural biology. J. Mol. Endocrinol. 2013, 51, T1–T21.
- Lamichane, S.; Dahal Lamichane, B.; Kwon, S.M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. Int. J. Mol. Sci. 2018, 19, 949.
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435.
- Liang, C.P.; Tall, A.R. Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in ob/ob mouse liver. J. Biol. Chem. 2001, 276, 49066–49076.
- You, M.; Considine, R.V.; Leone, T.C.; Kelly, D.P.; Crabb, D.W. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 2005, 42, 568–577.
- Zhou, Y.C.; Waxman, D.J. Cross-talk between janus kinase-signal transducer and activator of transcription (JAK-STAT) and peroxisome proliferator-activated receptor-alpha (PPARalpha) signaling pathways. Growth hormone inhibition of pparalpha transcriptional activity mediated by stat5b. J. Biol. Chem. 1999, 274, 2672–2681.
- Steineger, H.H.; Sorensen, H.N.; Tugwood, J.D.; Skrede, S.; Spydevold, O.; Gautvik, K.M. Dexamethasone and insulin demonstrate marked and opposite regulation of the steady-state mRNA level of the peroxisomal proliferator-activated receptor (PPAR) in hepatic cells. Hormonal modulation of fatty-acid-induced transcription. Eur. J. Biochem. 1994, 225, 967–974.
- Bishop-Bailey, D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br. J. Pharmacol. 2000, 129, 823–834.
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733.
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772.
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214.
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498.
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421.
- Reilly, S.M.; Lee, C.H. PPAR delta as a therapeutic target in metabolic disease. FEBS Lett. 2008, 582, 26–31.
- Usuda, D.; Kanda, T. Peroxisome proliferator-activated receptors for hypertension. World J. Cardiol. 2014, 6, 744–754.
- Azhar, S. Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease. Future Cardiol. 2010, 6, 657–691.
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566.
- Berger, J.P.; Akiyama, T.E.; Meinke, P.T. PPARs: Therapeutic targets for metabolic disease. Trends Pharmacol. Sci. 2005, 26, 244–251.
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747.
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271.
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830.
- Pehkonen, P.; Welter-Stahl, L.; Diwo, J.; Ryynanen, J.; Wienecke-Baldacchino, A.; Heikkinen, S.; Treuter, E.; Steffensen, K.R.; Carlberg, C. Genome-wide landscape of liver X receptor chromatin binding and gene regulation in human macrophages. BMC Genom. 2012, 13, 50.
- Yang, C.; McDonald, J.G.; Patel, A.; Zhang, Y.; Umetani, M.; Xu, F.; Westover, E.J.; Covey, D.F.; Mangelsdorf, D.J.; Cohen, J.C.; et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J. Biol. Chem. 2006, 281, 27816–27826.
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404.
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704.
- Yu, L.; York, J.; von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J. Biol. Chem. 2003, 278, 15565–15570.
- Tontonoz, P. Transcriptional and posttranscriptional control of cholesterol homeostasis by liver X receptors. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 129–137.
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838.
- Cha, J.Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751.
- Li, C.; Li, Y.; Gai, Z. Bile Acids and Farnesoid X Receptor: Novel Target for the Treatment of Diabetic Cardiomyopathy. Curr. Protein Pept. Sci. 2019, 20, 976–983.
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225.
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Pares, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751–761.e758.
- Xu, J.; Li, Y.; Chen, W.D.; Xu, Y.; Yin, L.; Ge, X.; Jadhav, K.; Adorini, L.; Zhang, Y. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology 2014, 59, 1761–1771.
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272.
- Sladek, F.M.; Zhong, W.M.; Lai, E.; Darnell, J.E., Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990, 4, 2353–2365.
- Chartier, F.L.; Bossu, J.P.; Laudet, V.; Fruchart, J.C.; Laine, B. Cloning and sequencing of cDNAs encoding the human hepatocyte nuclear factor 4 indicate the presence of two isoforms in human liver. Gene 1994, 147, 269–272.
- Hadzopoulou-Cladaras, M.; Kistanova, E.; Evagelopoulou, C.; Zeng, S.; Cladaras, C.; Ladias, J.A. Functional domains of the nuclear receptor hepatocyte nuclear factor 4. J. Biol. Chem. 1997, 272, 539–550.
- Dhe-Paganon, S.; Ottinger, E.A.; Nolte, R.T.; Eck, M.J.; Shoelson, S.E. Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proc. Natl. Acad. Sci. USA 1999, 96, 8378–8383.
- Lu, H. Crosstalk of HNF4alpha with extracellular and intracellular signaling pathways in the regulation of hepatic metabolism of drugs and lipids. Acta Pharm. Sin. B 2016, 6, 393–408.
- Yamagata, K.; Furuta, H.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Cox, N.J.; Fajans, S.S.; Signorini, S.; Stoffel, M.; Bell, G.I. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 1996, 384, 458–460.
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403.
- Zhou, W.; Hannoun, Z.; Jaffray, E.; Medine, C.N.; Black, J.R.; Greenhough, S.; Zhu, L.; Ross, J.A.; Forbes, S.; Wilmut, I.; et al. SUMOylation of HNF4alpha regulates protein stability and hepatocyte function. J. Cell Sci. 2012, 125, 3630–3635.
- Veto, B.; Bojcsuk, D.; Bacquet, C.; Kiss, J.; Sipeki, S.; Martin, L.; Buday, L.; Balint, B.L.; Aranyi, T. The transcriptional activity of hepatocyte nuclear factor 4 alpha is inhibited via phosphorylation by ERK1/2. PLoS ONE 2017, 12, e0172020.
- Chereji, R.V.; Morozov, A.V. Functional roles of nucleosome stability and dynamics. Brief Funct. Genom. 2015, 14, 50–60.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Sadakierska-Chudy, A.; Filip, M. A comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197.
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 2013, 61, 289–317.
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705.
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59.
- Dominy, J.E., Jr.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 2012, 48, 900–913.
- Tao, Y.; Wu, Q.; Guo, X.; Zhang, Z.; Shen, Y.; Wang, F. MBD5 regulates iron metabolism via methylation-independent genomic targeting of Fth1 through KAT2A in mice. Br. J. Haematol. 2014, 166, 279–291.
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407.
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577.
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959.
- Partanen, A.; Motoyama, J.; Hui, C.C. Developmentally regulated expression of the transcriptional cofactors/histone acetyltransferases CBP and p300 during mouse embryogenesis. Int. J. Dev. Biol. 1999, 43, 487–494.
- Arany, Z.; Sellers, W.R.; Livingston, D.M.; Eckner, R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 1994, 77, 799–800.
- Krebs, E.G. The Albert Lasker Medical Awards. Role of the cyclic AMP-dependent protein kinase in signal transduction. JAMA 1989, 262, 1815–1818.
- Zhou, X.Y.; Shibusawa, N.; Naik, K.; Porras, D.; Temple, K.; Ou, H.; Kaihara, K.; Roe, M.W.; Brady, M.J.; Wondisford, F.E. Insulin regulation of hepatic gluconeogenesis through phosphorylation of CREB-binding protein. Nat. Med. 2004, 10, 633–637.
- Liu, Y.; Dentin, R.; Chen, D.; Hedrick, S.; Ravnskjaer, K.; Schenk, S.; Milne, J.; Meyers, D.J.; Cole, P.; Yates, J., 3rd; et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 2008, 456, 269–273.
- Nakae, J.; Kitamura, T.; Silver, D.L.; Accili, D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Investig. 2001, 108, 1359–1367.
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.T.; Depinho, R.A.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311.
- Wondisford, A.R.; Xiong, L.; Chang, E.; Meng, S.; Meyers, D.J.; Li, M.; Cole, P.A.; He, L. Control of Foxo1 gene expression by co-activator P300. J. Biol. Chem. 2014, 289, 4326–4333.
- Hems, D.A.; Whitton, P.D.; Taylor, E.A. Glycogen synthesis in the perfused liver of the starved rat. Biochem. J. 1972, 129, 529–538.
- Newgard, C.B.; Moore, S.V.; Foster, D.W.; McGarry, J.D. Efficient hepatic glycogen synthesis in refeeding rats requires continued carbon flow through the gluconeogenic pathway. J. Biol. Chem. 1984, 259, 6958–6963.
- Shulman, G.I.; Rothman, D.L.; Smith, D.; Johnson, C.M.; Blair, J.B.; Shulman, R.G.; DeFronzo, R.A. Mechanism of liver glycogen repletion in vivo by nuclear magnetic resonance spectroscopy. J. Clin. Investig. 1985, 76, 1229–1236.
- Katz, J.; McGarry, J.D. The glucose paradox. Is glucose a substrate for liver metabolism? J. Clin. Investig. 1984, 74, 1901–1909.
- He, L.; Cao, J.; Meng, S.; Ma, A.; Radovick, S.; Wondisford, F.E. Activation of basal gluconeogenesis by coactivator p300 maintains hepatic glycogen storage. Mol. Endocrinol. 2013, 27, 1322–1332.
- Qiang, L.; Banks, A.S.; Accili, D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J. Biol. Chem. 2010, 285, 27396–27401.
- Ponugoti, B.; Kim, D.H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970.
- Kemper, J.K.; Xiao, Z.; Ponugoti, B.; Miao, J.; Fang, S.; Kanamaluru, D.; Tsang, S.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 2009, 10, 392–404.
- Conkright, M.D.; Canettieri, G.; Screaton, R.; Guzman, E.; Miraglia, L.; Hogenesch, J.B.; Montminy, M. TORCs: Transducers of regulated CREB activity. Mol. Cell 2003, 12, 413–423.
- Kobayashi, M.; Deguchi, Y.; Nozaki, Y.; Higami, Y. Contribution of PGC-1alpha to Obesity- and Caloric Restriction-Related Physiological Changes in White Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 6025.
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138.
- Estall, J.L.; Kahn, M.; Cooper, M.P.; Fisher, F.M.; Wu, M.K.; Laznik, D.; Qu, L.; Cohen, D.E.; Shulman, G.I.; Spiegelman, B.M. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes 2009, 58, 1499–1508.
- Corona, J.C.; Duchen, M.R. PPARgamma and PGC-1alpha as therapeutic targets in Parkinson’s. Neurochem. Res. 2015, 40, 308–316.
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555.
- Besse-Patin, A.; Jeromson, S.; Levesque-Damphousse, P.; Secco, B.; Laplante, M.; Estall, J.L. PGC1A regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc. Natl. Acad. Sci. USA 2019, 116, 4285–4290.
- Li, S.; Liu, C.; Li, N.; Hao, T.; Han, T.; Hill, D.E.; Vidal, M.; Lin, J.D. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 2008, 8, 105–117.
- Morris, E.M.; Meers, G.M.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1alpha overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992.
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028.
- Shapiro, M.J.; Shapiro, V.S. Transcriptional repressors, corepressors and chromatin modifying enzymes in T cell development. Cytokine 2011, 53, 271–281.
- Ahmad, K.F.; Melnick, A.; Lax, S.; Bouchard, D.; Liu, J.; Kiang, C.L.; Mayer, S.; Takahashi, S.; Licht, J.D.; Prive, G.G. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol. Cell 2003, 12, 1551–1564.
- Wu, X.; Li, H.; Park, E.J.; Chen, J.D. SMRTE inhibits MEF2C transcriptional activation by targeting HDAC4 and 5 to nuclear domains. J. Biol. Chem. 2001, 276, 24177–24185.
- Jayne, S.; Zwartjes, C.G.; van Schaik, F.M.; Timmers, H.T. Involvement of the SMRT/NCoR-HDAC3 complex in transcriptional repression by the CNOT2 subunit of the human Ccr4-Not complex. Biochem. J. 2006, 398, 461–467.
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835.
- Ye, J. Improving insulin sensitivity with HDAC inhibitor. Diabetes 2013, 62, 685–687.