Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Allogeneic cell therapies, defined by genetically mismatched transplantation, have the potential to become a cost-effective solution for cell-based cancer immunotherapy. This type of therapy is often accompanied by the development of graft-versus-host disease (GvHD), induced by the mismatched major histocompatibility complex (MHC) between healthy donors and recipients, leading to severe complications and death.
- graft-versus-host disease (GvHD)
- GvHD modulation
- innate T cell
- mucosal-associated invariant T (MAIT) cell
1. Introduction
Graft-versus-host disease (GvHD) is a common complication associated with allogeneic transplantation such as allogeneic hematopoietic stem cell transplantation (allo HSCT) and chimeric antigen receptor T (CAR-T)-cell therapies. With their highly variable αβ T-cell receptors (TCRs), donor T cells bind with major histocompatibility complex (MHC) class I and II molecules widely expressed on recipient tissue cells [1]. When the TCR recognizes the MHC as foreign, donor T cells initiate the immune response and attack recipient cells, causing an alloreaction leading to GvHD. Moreover, in adoptive cell therapies without preconditioning, recipient T cells can also recognize mismatched donor MHC molecules and attack transplanted cells. However, even with MHC-matched donors such as siblings, patients develop acute and chronic GvHD in 25–40% and 40–60% of the cases, respectively [2]. Under these circumstances, GvHD is induced by the disparity between donor and recipient minor histocompatibility antigens (miHAs), peptides bound to MHC molecules that are capable of triggering a donor T cell immune response [3].
Mediators of GvHD include several populations of T cells, such as CD4+ T cells, CD8+ T cells, and CD4/CD8 double-positive populations. These T-cell populations induce GvHD via a perforin-dependent pathway [4] and secretion of interferons (IFNs) [5]. Different memory T-cell subsets have varying abilities to mediate GvHD based on their phenotypes. For instance, whereas CD62L+ naïve T cells and CD44+CD62L+ central memory T cells are capable of inducing GvHD due to their high alloreactivity [6,7], CD62L- memory T cells do not cause GvHD in general [8,9]. In the case of Th17 cells, studies have indicated that GvHD progression is exacerbated after polarization [10] and that in vitro differentiation leads to severe GvHD with pulmonary damage [11]. Furthermore, CD3+CD4+CD25hiFoxP3+ regulatory T cells (Tregs) alleviate GvHD by directly regulating effector T cell function via the secretion of inhibitory cytokines [12].
Current therapies against GvHD work by manipulating mediators of the disease, the most common mediator being T cells. To address this, the most common treatment for GvHD is T-cell depletion (TCD), allowing accurate elimination of specific T-cell subsets, usually via magnetic-associated cell sorting [13]. Studies on CD34+ TCD in CAR-T-cell transplantation indicate that alloreactive T cells from the graft are effectively depleted and the incidence of GvHD and relapse is significantly decreased [14,15]. Manipulating other mediators of alloreactivity can also be useful, such as expanding Tregs to alleviate GvHD or recruiting NK and gamma delta T cells to restore immunity following depletion [16,17], considering that TCD may lead to diminished reactivity and efficacy of the grafted effector cells [13]. In addition, myeloid-derived suppressor cells (MDSCs), which are formed under chronic inflammation or infection and stimulated by signals including macrophage (M-)/granulocyte (G-)/granulocyte–macrophage (GM-) colony-stimulating factors (CSFs) and proinflammatory cytokines such as IFN-γ, IL-4, IL-6, and IL-23 [18,19], play a significant role in the treatment of GvHD with their immunosuppressive function, shielding grafted cells from alloreactivity. MDSCs are also capable of assisting Treg expansion and inhibiting the proliferation of T cells through cytokine secretion, suppressing the inflammation induced by GvHD [19].
One limitation associated with the depletion-based therapies for GvHD described above is that they focus on eradicating reactive T cells rather than addressing MHC recognition, severely limiting the efficacy of the initial adoptive cell transfer. A potential solution being explored is harnessing the MHC-independent properties of innate lymphocytes to limit GvHD responses in patients. With their semi-invariant chains and restricted number of α/β/γ chains [20], innate T cells induce cytotoxicity through MHC-independent mechanisms, enabling the transplantation of allogeneic effector cells without the risk of GvHD. In addition to their MHC-independent TCR activation, each of these innate T cells also has a unique mechanism to minimize GvHD (Figure 1).
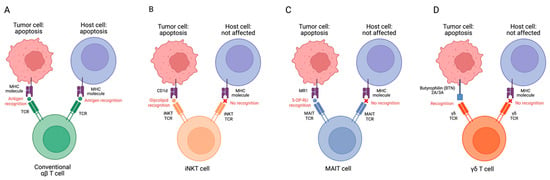
Figure 1. Targeting tumor and host cells by conventional αβ T (A), iNKT (B), MAIT (C), and γδ T cells (D). MHC–antigen–TCR interactions allow T cells to recognize target cells. MHC molecules on host cells could be recognized by conventional T cell TCRs after allogeneic cell infusion, resulting in GvHD. However, innate T cell TCRs do not recognize mismatched MHCs and protein antigens, therefore, these cells have no GVHD risk. MR1, major histocompatibility complex, class I-related protein; 5-OP-RU, 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil.
2. MAIT Cell Modulation of GvHD
MAIT cells are a subset of innate T cells found in the blood, liver, and the epithelial layers of the lung and the respiratory and GI tracts [49,50]. These cells are quiescent until activated by microbial infections. MAIT cells exhibit a CD161hi phenotype and have a semi-invariant TCR composed of an invariant α chain and a β chain from a variant β repertoire [26]. MAIT cells have several mechanisms for activation, including TCR-dependent and -independent pathways. The TCR-dependent pathway involves TCR recognition of riboflavin-derived antigens presented by MR1, an MHC class I-like molecule for MAIT cell activation [51]. The primary ligand for MR1 is 5-(2-oxopropylideneamino)-6-d-ribitylaminouracil (5-OP-RU), an intermediate of riboflavin biosynthesis by microbial pathogens [52]. Studies have demonstrated the effector activity of MAIT cells in vitro is significantly enhanced with the administration of synthetic 5-OP-RU, indicating its potential role in mediating MAIT populations [48]. The TCR-independent mechanism involves inflammatory cytokines including IL-7, IL-12, and IL 18 [50]. Upon activation, MAIT cells proliferate, accumulate in vivo, initiate cytotoxic function via perforin and granzymes, and secrete pro-inflammatory cytokines (e.g., IFN-γ, TNF-α, IL-15, IL-17, and GM-CSF) to recruit circulating effector cells in vivo [53].
The unique biology of MAIT cells allows them to minimize the occurrence of GvHD from MHC mismatch. MAIT cells have been shown to be crucial in both acute and chronic GvHD, particularly when it comes to complications of the gut and skin, where MAIT cells reside. In patients with allo HSCT, Gao et al. found that there was a higher risk of acute GvHD in grafts with lower frequencies of MAIT cells [21,25]. Acute GvHD may also be influenced by the intestinal microbiota: acute GvHD is more likely to develop when there are more non-riboflavin pathways [22], further providing evidence that MAIT cell activation plays a key role in GvHD modulation.
MAIT cells in allografts influence chronic GvHD as well. Although the long-term reconstitution of MAIT cells after allograft transplantation may be affected by various factors including the diversity of gut microbiota, different donor sources, and MAIT cell number/type in the transplanted tissue [24], the early reconstitution is influenced by MAIT cell proliferation after transplantation [26]. Higher MAIT cell counts in allografts reduce the risk of poor MAIT reconstitution and, consequently, the incidence of GvHD [24]. Along with clinical results, in vitro studies of CAR-MAIT cells showed that these cells exhibit high efficacy and safety against tumors, minimizing GvHD in allogeneic cell-based therapy [48]. The CAR-MAIT cell TCR identifies high levels of MR1 molecules on myeloid-cell-derived APCs, which have been found to exacerbate acute and chronic donor T-cell-induced GvHD; hence, CAR-MAIT cells may remove these myeloid APCs and diminish GvHD [48,54,55].
3. iNKT Cells Modulation of GvHD
iNKT cells are a subset of T cells that play a role in bridging the adaptive and innate immune systems with a variety of mechanisms [56,57]. As an innate lymphocyte population, iNKT cells express the semi-invariant Vα24Jα18 TCR in humans, paired with a limited Vβ chain [42,58,59,60]. Similar to other innate immune cells, iNKT cells exhibit less specificity and a quicker activation than adaptive lymphocytes [61]. iNKT cells recognize lipid antigens presented on CD1d [57,59], a non-polymorphic MHC class I-like molecule [42,57,59,61]. Due to the degree of conservation in the canonical TCR and CD1d molecules, interspecies cross-reactivity is possible [62]. For instance, mouse iNKT cells are capable of reacting with human CD1d molecules and vice versa, demonstrating iNKT cells’ potentially broad role in the immune system [62].
As mentioned before, one distinguishing characteristic of iNKT cells is that they recognize glycolipids presented on CD1d [60]. The majority of the known antigens are composed of a similar structure of a lipid tail buried into the CD1d surface protein and a sugar head group emerging to make contact with the iNKT TCR [62]. The glycolipid α-galactosylceramide (α-GalCer) was the first discovered crucial activator for iNKT cells [57,63]. Although iNKT cells possess both TCR α and β chains, evidence supports that recognition of CD1d is carried out via the TCRα chain with four essential amino acids: Asp94, Arg95, Gly96, and Ser97. The TCRβ chain is shown to not be involved in the binding process [64,65,66,67]. Although binding with cognate antigens, such as microbial glycolipids, can directly stimulate iNKT cells, indirect activation of iNKT cells occurs through two primary methods for pathogens that lack cognate antigens: partial CD1d-TCR-binding-dependent activation combined with antigen presenting cell (APC) stimulation or CD1d-independent activation [62].
iNKT cells have been shown to play a role in modulating the GvHD response in transplant patients [27,32,33,34,35,38,56,68]. With reduced intensity conditioning (RIC), total lymphoid irradiation (TLI), and antithymocyte globulin (ATG), iNKT cells are able to alleviate GvHD in transplant patients [27,41,69]. Moreover, host iNKT cells trigger the expansion of Tregs, which play an essential role in the immunosuppression required to avoid GvHD [31]. The severity of GvHD in humans has been found to be correlated with the persistence of iNKT. [29,70,71,72,73,74]. The incidence of GvHD is also reduced in grafts that include more donor iNKT cells [56]. For instance, one study has demonstrated that the number of iNKT cells in the cryopreserved graft significantly increased GvHD-free progression-free survival (GPFS) among patients undergoing peripheral blood stem cell transplantation (PBSCT) by an average of two years [28].
Because of iNKT cells’ potential in reducing GvHD, the therapeutic potential of iNKT cells is being increasingly studied. One method to enhance the therapeutic potential of iNKT cells is through ex vivo expansion via single antigenic stimulation [40]. In the study conducted by Trujjilo-Ocampo et. al., following enrichment from peripheral blood mononuclear cells (PBMCs), iNKT cells are cultured with antigen-presenting dendritic cells for two weeks with agonist glycolipids such as α-GalCer [40]. Expanded iNKT cells express high levels of CD4 and alleviate xenograft GvHD, as evidenced by a higher survival rate for the iNKT-treated mice, as well as significantly less severe GvHD features in the skin, small intestine, liver, and lung compared with those in the PBMC-only-treated mice. The suppression of the proliferation of conventional T cells was observed as well, which might be the consequence of strong TCR-mediated activation of responder T cells or the high ratio of responder T cells to iNKT cells [40].
4. γδ T Cell Modulation of GvHD
γδ T cells are a highly heterogeneous population of T lymphocytes that exhibit qualities of both innate and adaptive immune cells. They are defined by a γδ TCR unique from the conventional αβ T cells in their antigen recognition, anatomical distribution, and killing mechanisms [76]. γδ T cells normally constitute between 1–10% of the total T cells in the human body and operate independently of HLA recognition during the initial phases of the immune response [43]. The γδ TCR loci are the T-cell receptor gamma (TRG) and the T-cell receptor delta (TRD) loci, and rearrangement is dependent on recombination activating genes (RAG). These loci are much more restricted than in αβ T cells, with only six functional TRG V segment genes and eight functional TRD genes (compared with 70 Vα and 52 Vβ genes) [77]. The TCR composition largely determines the localization of the T cell, with Vδ1, Vδ3, or Vδ5 TCRs localizing in epithelial tissues and Vγ9Vδ2 more commonly circulating in the peripheral blood [78,79]. The vast majority of γδ T cells do not express CD4 or CD8 coreceptors but rather express several NK receptors for NK-like function (e.g., CD16, NKG2D, MIC-A, MIC-B, and UL16) [80,81].
Whereas αβ T cells migrate to the thymus, many γδ T migrate to tissues such as the epidermis, dermis, intestines, or uterus. In addition, unlike conventional αβ T cells, γδ T cells recognize molecules independently of MHC [76]. TCR composition and coreceptor expression largely dictate the physiological distribution and antigen specificity of different γδ T cell subtypes. The most common peripherally circulating γδ T cells, Vγ9Vδ2 γδ T cells, recognize phosphoantigens produced by microbes or malignancies, and tissue-localized Vδ1 and Vδ3 γδ T cells recognize molecules presented by the CD1 family of surface receptors [77]. Vγ8Vδ3 peripheral blood γδ T cells recognize annexin A2, and Vγ4Vδ5 γδ T cells recognize endothelial protein C receptors in the peripheral blood [82,83].
Effector function in γδ T cells is activated through various means, including secreting regulatory cytokines, releasing perforin, granzymes, and IFN-γ, and antibody-dependent cellular cytotoxicity (ADCC) via CD16 [84]. Effector function in γδ T cells may vary depending on the individual cell’s niche, with intestinal-resident γδ T cells secreting keratinocyte growth factor for epithelial homeostasis [77]. Other effector functions may be induced either dependent or independent on TCR recognition. For example, IL-1β- and IL-23- induced cytokine production allows γδ T cells to secrete IL-17 and other regulatory cytokines in humans [85]. Furthermore, TCR- and NK-marker activation may enable γδ T cells to enter a pro-inflammatory state, secrete IFN-γ, TNF-α, and IL-17, and induce cells to enter a state of antigen presentation, thereby promoting the adaptive immune response in CD4+ and CD8+ T cells [10]. In this manner, γδ T cells serve a role in bridging the innate and adaptive immune responses, activating with and without antigen stimulation.
The nature of γδ T cells’ MHC-independent activation suggests that they may play a vital role in mediating GvHD for allogeneic cell therapies. In patients with allogeneic stem cell transplantation for hematological malignancies, higher concentrations of γδ T cells about two months after transplantation correlated with improved overall survival and relapse-free survival. Moreover, the risk of acute GvHD one month after transplantation was greatly reduced for patients with higher levels of γδ T cells [43]. Speculatively, allogeneic γδ T cells may alleviate GvHD by immunoregulatory actions such as IL-4 secretion or cytotoxic effects on CD277-expressing APCs [48,86]. These data suggest a role for these lymphocytes in protecting against tumor cells and against alloreactivity.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24044084
This entry is offline, you can click here to edit this entry!