2. Current Status of LGGs in the WHO Classification
Historically, gliomas were classified primarily based on their histologic attributes [
1,
3]. This practice continued until the 2007 WHO classification, which recognized seven different types of gliomas, based on differentiation along astrocytic and/or oligodendroglial lineages [
10]. Further prognostic entities were later defined based on the histologic grading, with cellular features of mitoses and necroses associated with both higher grades and worse prognosis [
10]. However, this classification system suffered from significant intra-observer and inter-observer variability, along with a lack of clarity regarding reproducible methods.
With advances in molecular analysis, glioma classification has undergone a paradigm shift, with significant molecular heterogeneity reported among each histologic type of glioma [
1,
6,
11,
12]. One such seminal advance was the discovery of mutations in the isocitrate dehydrogenase (IDH) 1 and 2 genes, with IDH1/2 mutations identified in over 70% of LGGs [
13]. Furthermore, IDH1/2-mutant (IDHmt) tumors were found to have a demonstrably better prognosis than IDH1/2-wild type (IDHwt). In 2015, a study utilizing The Cancer Genome Atlas (TCGA) analyzed 293 LGGs and identified an additional molecular marker—the loss of chromosomes 1p and 19q—allowing subclassification into three prognostically distinct groups. Arranged from best to worst prognosis, LGGS can be fundamentally ordered into (A) IDH-mutant (IDHmt) LGGs with 1p/19q chromosomal codeletion, e.g., oligodendrogliomas, which are associated with gene mutations of Telomerase Reverse Transcriptase (TERT); (B) IDHmt LGGs without 1p/19q chromosomal codeletion, e.g., astrocytomas that are typically associated with mutations in Tumor Protein 53 (TP53) and ATP-Dependent Helicase ATRX (ATRX); and (C) IDH wild-type (IDHwt) LGGs [
14]. Subsequent studies elucidated genetic signatures unique to each of these three groups [
15,
16].
Recognizing these advances, the WHO 2016 classification of gliomas emerged, which utilized a combination of histologic and molecular signatures for classification [
17]. Here, six separate entities of glioma were identified, each with a unique molecular signature. While this was a welcome step, one persistent limitation was the continued reliance on ‘brisk’ mitotic activity to distinguish Grade 3 from Grade 2 gliomas, requiring subjective counting of specimens, something that was compounded by the fact that mitotic activity had little significance in IDHmt LGGs [
18].
The most recent, fifth edition of the WHO Classification of Tumors of the Central Nervous System (WHO CNS5) took this one step further by incorporating the recommendations from the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) [
14,
19,
20,
21,
22], along with the landmark DNA methylation-based classification of CNS tumors published in Nature [
12]. The WHO CNS5 uses an integrated histo-molecular assessment, prioritizing genetic and molecular alterations, which were emphasized for several tumor types [
5]. A summary of the view of the WHO CNS5 has been provided in
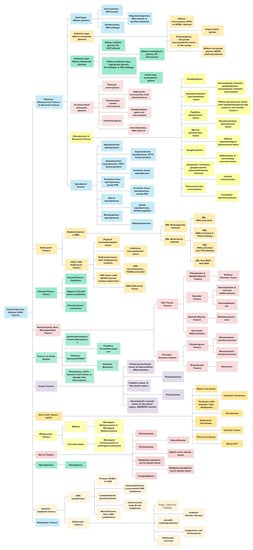
Figure 1.
Figure 1. A summary view of the World Health Organization (WHO) 2021 classification of central nervous system (CNS) tumors. This original figure has been created by authors using data available from the WHO CNS5 publication.
WHO CNS5 utilizes a hybrid approach with regard to tumor grouping [
23]. While some tumor groups still find a lack of utilization for any molecular testing requirements, such as meningiomas, several new types, and subtypes are primarily characterized by molecular features such as medulloblastoma and ependymomas [
5]. Gliomas currently fall under the group of “Gliomas, Glioneuronal and Neuronal Tumors”. The grading of gliomas, now done using WHO Grade 1–4 instead of Grade I–IV, is to be based on a combination of histologic and molecular features [
5]. Gliomas have also been separated into pediatric-type and adult-type, thus reorganizing and grouping entities with common genetic alterations. Gliomas were also rearranged, accounting for their prevalent genetic mutations, especially IDH 1/2 mutation (better prognosis), 1p/19q codeletion (better prognosis), and CDKN2A/B homozygous deletion (worse prognosis). Grading is now to be done within individual tumor types instead of across tumor types. Perhaps the most landmark change for clinicians was the change in the classification of glioblastomas (GBMs). As per WHO CNS5, GBM includes only IDH-wild type entities, while previously GBMs included both IDHmt (10%) and IDHwt (90%) [
24].
As per WHO CNS5, diffuse astrocytic tumors can now be classified as Grade 2 (i.e., LGG), Grade 3, or Grade 4, the latter two being high-grade gliomas (HGGs). Diffuse astrocytic tumors with IDHwt, i.e., baseline more aggressive than IDHmt, that lack GBM-specific histology but have at least one of three particular genetic alterations would also be classified as GBMs [
5]. These specific alterations are (1) TERT promoter mutations (TERT-pmt), (2) EGFR gene amplifications, and/or (3) loss of chromosome 10 (+7/−10) [
5,
23]. On the other hand, IDHmt astrocytomas with CDKN2A/B homozygous deletions and related alterations can now be classified as WHO Grade 4, even if histologically lacking necroses or microvascular proliferation [
5]. Thus, IDH mutation testing has become a key requirement for appropriate classification into LGG or HGG [
23]. The characterization of methylomic attributes was added to diagnostic criteria, albeit as “desirable characteristics”, acknowledging the general inaccessibility of these tools [
25].
While recognizing newer or updated entities in the new classification, it is also essential to note that low-grade gliomas (LGG), in particular astrocytomas, can transform into higher-grade tumors or display more aggressive behavior after some time [
2]. Nearly 70% of diffuse LGGs transform into a higher-grade type [
26,
27]. This is likely the result of the gradual accumulation of genetic and epigenetic alterations, which together allow cellular replication to take place in an unrestrained fashion. Epigenetic alterations in cancer cells have been demonstrated to increase genomic fragility, increase angiogenic capabilities, decrease the attribute of cellular adhesion, permit entry into the cell cycle, help avoid apoptosis and lead to defects in DNA repair, as further examined below [
28].
3. Overview of DNA Methylation and Demethylation
The importance of epigenetic processes in clinical neurosciences may be amply demonstrated in the role of DNA methylation patterns in the physiological regulation of differentiation, in particular, through cellular, spatial, and temporal specificities [
29,
30]. Notably, epigenetic deregulation has also been included among the updated hallmarks of cancer [
31,
32]. In the cancer cell, it acts in both a standalone fashion and synergistically with other genetic changes in driving neoplastic evolution [
29,
30,
31,
32]. However, despite substantial advances in the understanding and the utility of investigating methylomics of various malignancies, considerably less headway has been made in the clinical utilization of epigenetics in brain tumors, especially in less aggressive tumors such as low-grade gliomas [
30]. DNA methylation has been the most widely studied and most clinically explored epigenetic change in gliomas [
28].
The cellular DNA, including that of the cancer cell, is constructed out of four elements (DNA bases), namely, adenine (A), thymine (T), guanine (G), and cytosine (C). While adenine and guanine are purines, thymine and cytosine are pyrimidines. Base pairing occurs between AT and GC, but a methylated cytosine, with its corresponding CG base pairing, may undergo deamination to form a thymine.
DNA methylation is a process by which methyl (CH3-) groups are added to the DNA bases, to allow for an additional layer of regulation of gene expression. This modification can change the activity of a DNA segment without changing the underlying sequence. DNA methylation typically occurs on cytosine bases, leading to the formation of 5-methylcytosine, often called the ‘fifth DNA base’. It is estimated that 3–6% of cytosine bases in human cells carry methyl groups [
28], where it is especially predominant in repetitive genomic sequences. The constant methylation status of these sequences has been reported to potentially play a role in the routine upkeep of healthy cells by averting chromosomal instability, translocations, and genetic disruptions. The latter, which may occur through the reactivation of certain transposon-derived sequences that have self-propagation and random site insertion properties, is prevented by hypermethylation [
28,
33]. Additionally, DNA methylation is one of the most reliable means to transmit epigenetic information across cellular replication [
34,
35,
36]. Thus, maintaining the integrity of DNA methylation patterns is essential for proper cellular function, and disruptions to this process can have significant effects on health and disease.
Because cytosine is typically paired with guanine, a DNA sequence where several methylated cytosine and guanine pairs come together are known as ‘CpG or CG Islands’, where the highest amount of methylation is present in the genome [
37]. CpG islands can be found throughout the genome, and their exact location and frequency can vary depending on the organism and the specific region of the genome [
37]. CpG islands frequently occur near the 5′ end of genes (~70%) that contain DNA sequences corresponding to the promoter, untranslated region (5′-UTR), and exon 1. Unmethylated CpG sites permit the related sequences to be expressed when the required transcriptional activators are available [
28,
38,
39].
The process of DNA methylation is carried out by DNA methyltransferases (DNMT), which transfer a methyl group from S-adenosyl methionine (SAM), a carrier molecule, to the DNA molecule, resulting in the addition of a methyl group to the cytosine base. While several of these enzymes exist, all of them utilize SAM as the carrier molecule. The proteins encoded by the
DNMT3 gene and its variants (
DNMT3A,
DNMT3B, regulatory
DNMT3L) preferentially methylate unmethylated DNA strands and thus carry out a major part of de novo methylation [
35]. Meanwhile, the proteins encoded by the
DNMT1 gene methylate DNA whose single strand has already been methylated (hemimethylated DNA) in a preferential fashion [
35]. This permits it to maintain the methylation patterns across cellular replication [
36,
40].
To ensure the reliability of DNA methylation, cells have several mechanisms in place to monitor and repair methylation patterns. For example, enzymes of the Ten-Eleven Translocases (TET) family (
TET1,
TET2,
TET3), can remove methyl groups from DNA, and help reverse the de novo methylation process, while other enzymes can recognize and repair damaged or improperly methylated DNA. TET enzymes, which are α-ketoglutarate-dependent dioxygenases, convert 5-methylCytosine (5mC) to 5-hydroxymethylCytosine (5hmC), 5-formylCytosine (5fC), and 5-carboxylCytosine (5caC) in a stepwise fashion [
41,
42], as part of the normal cytosine methylation cycle. The 5-carboxylcytosine is later removed by the human thymine-DNA glycosylase (hTDG) enzyme, in a process exemplifying “active DNA demethylation” [
43,
44]. This is immediately followed by the insertion of an unmethylated cytosine residue at the excision site, carried out by the DNA Base Excision Repair (BER) system [
45]. The TET-hTDG-BER system is known to ensure that cells can actively and rapidly demethylate specific loci in response to environmental changes, such as cellular stressors. This active demethylation is in contrast to the passive demethylation process which occurs in locations where DNMT1 is not present to methylate DNA during replication [
46]. Additionally, 5-hmC, by itself, has been hypothesized to play a role in the regulation of gene expression, given that it is noted to be present in both tissue-specific gene bodies and DNA enhancers, the latter being short regulatory sequences where transcription factors bind. Thus, dysregulation of this tightly controlled active methylation and active demethylation in healthy cells leads to errors that eventually permit the hallmark neoplastic features to manifest [
32]. Efforts are underway to generate genome-wide 5-hmC profiles (tissue maps) of cells in various tumors [
47].
4. DNA Methylation in Low-Grade Gliomas
The utility of studying DNA methylation was first identified in glioblastoma, due to its aggressiveness and poor prognosis. While such studies have begun to include low-grade gliomas (LGGs) as well, literature specific to LGGs remains scarce (Persico et al., 2022), even though there is wide recognition that DNA methylation is likely to play a key role in the next frontier of oncology diagnostics and therapeutics [
49,
50].
Fundamentally, methylation of a locus typically results in the repression of its expression level, which can then affect the expression level of other genes that are downstream targets. Methylated DNA sequences are less accessible to the cellular machinery that reads the genetic code. For example, if the locus has elements that repress expression (e.g., 5′ regulatory region) of the associated gene(s) (e.g., a DNA damage repair gene), then the methylated locus would become silenced, leading to an increase in gene expression of the associated gene (in this case higher production of DNA damage repair proteins).
In general, while cancer cells undergo a global loss of DNA methylation, CpG islands of tumor-suppressor genes (TSGs) undergo preferential hypermethylation [
28]. The epigenetic silencing of TSGs permits the cancer cell to evade pro-apoptotic changes, proceed with unrestrained cellular replication, display angiogenesis and reduce cellular adhesion, amongst other mechanisms, thus contributing to the classically described hallmarks of cancer cells [
28,
35,
51]. These unique DNA methylation changes are also accompanied by histone modifications, another epigenetic alteration that permits further silencing of TSGs and increased expression of oncogenes [
52,
53,
54], as discussed later in the text. Hypermethylation of tumor suppressor genes is increasingly being explored as a prognostic marker in low-grade gliomas, for instance, testing for MGMT methylation status to predict response to chemotherapy [
55]. O6-methylguanine-DNA methyltransferase (
MGMT) is a protein involved in DNA repair. When the
MGMT gene locus become methylated (i.e., hypermethylated), the amount of DNA repair across the genome reduces, leading to increased sensitivity to cytotoxic medications, making the tumor more responsive to chemotherapy [
33]. Therefore, in gliomas,
MGMT hypermethylation is associated with a better response to temozolomide, a DNA alkylating agent.
MGMT promoter hypermethylation is being increasingly explored as a clinical target in LGGs. It has been recently reported to be a predictor of hypermutation in LGGs at the time of recurrence. Mathur et al. demonstrated in 2020 that methylation-based silencing of
MGMT expression enhances mutagenic processes caused by temozolomide in LGGs, thus leading to the development of hypermutation in these tumors. Further, analysis of DNA methylome of genes involved in DNA damage repair in the EORTC 22033 trial cohort has demonstrated that patients having a high MGMT-STP27 score, which measures methylation status, prognosticates those patients of IDHmt LGGs who are most likely to benefit from temozolomide chemotherapy [
56]. Meanwhile, work from UCSF has demonstrated that temozolomide positively selects for tumor cells with
MGMT hypermethylation in patients with LGGs lacking DNA mismatch repair (MMR) [
57]. Given these and similar findings from the literature,
MGMT promoter methylation is likely to serve as a useful biomarker for predicting response to therapy and risk of hypermutation at recurrence [
56,
57,
58].
In addition to the involvement of DNA methylation in cellular processes in LGGs, errors in DNA methylation also predispose to mutations. Compared to cytosine (C), methylated cytosine residues (mC) are more prone to deamination, i.e., loss of the amine (-NH2) group, forming thymine residues, which are less likely to be repaired accurately [
45]. This mutational event then changes the DNA sequence, which is the primary driver of the sequence of corresponding messenger RNA, leading to abnormalities in structure, quantity, or function in subsequent protein synthesis. Thus, ‘CpG Islands’ are more prone to mutations than human DNA sequences in general. One pertinent example is the glioma CpG island methylator phenotype (G-CIMP), a pattern of genetic changes that includes MGMT methylation, which is often associated with the presence of
IDH1 or
IDH2 gene mutations. G-CIMP, while quite underexplored in LGGs, likely represents a major avenue for future research given that Grade 2 astrocytoma (IDHmt) and oligodendroglioma (IDHmt, 1p/19q codeletion) are both characteristically associated with G-CIMP. This attribute gains importance given that, amongst WHO Grade 2/3 astrocytomas, oligodendrogliomas, and glioblastomas developing from these lower grade entities, IDH1 mutation occurs at codon number 132 in over two-thirds of these, with IDH2 mutations occurring in 6% of them [
13]. Given that MGMT resides on chromosome 10, it has been reported that compared to GBM, where at least one copy of chromosome 10 is lost, IDHmt lower-grade gliomas do not lose either copy. Thus, sufficient silencing of the MGMT gene may not occur in these IDHmt gliomas, leading to MGMT expression, followed by remnant capacity for DNA repair. This is the likely cause behind the resistance of IDHmt gliomas to temozolomide chemotherapy, compared to GBM [
45]. Additionally, the deletion of 1p36 has been demonstrated to occur in nearly 73% of oligodendrogliomas and 18% of astrocytomas, while the deletion of 19q13.3 chromosome has been found to occur in 73% of oligodendrogliomas and 38% of astrocytomas. 1p/19q-codeletion has been demonstrated to occur in nearly 64% of oligodendrogliomas and 11% of astrocytomas [
59,
60].
Additionally, methylation is known to alter the overall 3-dimensional organization of chromatin protein used for DNA compaction. Chromatin consists of loops or topology-associated domains (TADs), which are normally conserved and maintained across cells [
45]. The architecture of TADs has been demonstrated to be disturbed in IDHmt gliomas, causing excessive oncogene and anti-apoptotic factor expression [
61,
62]. One example is the Cohesin and CCCTC-binding factor (CTCF), whose alteration affects the organization of TADs [
45].
DNA methylation has also recently been implicated in the functioning of the Telomerase Reverse Transcriptase (TERT) gene. TERT-promoter mutations (TERT-pmt) are known to be amongst the most common and the earliest mutations in the most invasive gliomas [
63,
64,
65,
66,
67]. TERT mutations have been reported to be closely associated with
IDH1/2 mutations and 1p/19q-codeletion in oligodendroglioma, but less well correlated in astrocytomas [
68,
69]. It has been hypothesized that TERT promoter mutations enhance the neoplastic potential of tumors with low rates of self-renewal, such as low-grade gliomas [
70]. Where methylation additionally plays a role is in the regulation of the TERT gene, whose promoter region has elements called “GC boxes”. These GC-base pair-rich DNA sequences preferentially bind to the transcriptional activator SP1, leading to increased gene expression. These GC boxes are closely regulated through DNA methylation [
71]. Furthermore, hypermethylation of the TERT promoter region has been demonstrated to be one factor behind the dysregulation of TERT function in cancer cells [
72,
73,
74]. Uniquely, TERT hypermethylated oncological region (THOR), a 433-bp sequence, has been reported to be a region where methylation leads to increased transcriptional TERT activity. It is situated just upstream of the TERT promoter region and contains 52 CpG sites. THOR hypermethylation has been demonstrated to play a role in the pathogenesis and/or outcomes of several pediatric brain tumors, including gliomas [
75,
76,
77].
DNA methylation, within the context of low-grade gliomas, also plays a role in the regulation of the ADP-ribosylation factor-like (ARL) family of genes. The ADP-ribosylation factor (ARF) family of proteins, a part of the RAS superfamily, had been previously demonstrated to play a part in the pathogenesis of both glioblastoma and lower-grade gliomas [
78,
79,
80]. Utilizing the TCGA database, Tan et al. recently identified low expression of ARL9 mRNA, along with ARL9 hypermethylation, which had hitherto been unexplored in LGGs, as positive prognostic factors in LGG [
81]. The ARL9 protein expression was reported as correlating with CD8 T-cells in the LGG tissue, indicating the role of ARL9 methylation in tumor immune infiltration [
81].
Broad prognostic signatures based on epigenetics have been very recently developed for low-grade gliomas. A two-CpG site DNA methylation signature (GALNT9 and TMTC4, both of whose expressions are highly dependent on methylation) has been recently identified that correlated highly with prognosis, regardless of the age, WHO grade, family history of cancer, and IDH mutation status [
82]. Similarly, three methylation-driven genes (ARL9, CMYA5, STEAP3) have been recently identified as independent prognostic factors for survival in LGGs [
83].
Overall, DNA methylation is an important mechanism for regulating gene expression in cancer cells, including LGGs, through several pathways. Alterations in DNA methylation lead to changes in gene expression that can result in neoplastic processes. The precise pattern of DNA methylation likely varies between cells of different grades and types of LGGs, being influenced by several factors, most of which are under investigation.