Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cardiac hepatopathy refers to acute or chronic liver damage caused by cardiac dysfunction in the absence of any other possible causative reasons of liver injury. There is a large number of evidence of the fact that cardiac hepatopathy is associated with poor clinical outcomes in patients with acute or actually decompensated heart failure (HF).
- heart failure
- cardiac hepatopathy
- inflammation
- oxidative stress
- biomarkers
1. Introduction
The number of new cases of heart failure (HF) is steadily increasing worldwide. The prevalence of HF with preserved ejection fraction (HFpEF) is currently higher than that of HF with reduced (HFrEF) and mildly reduced (HFmrEF) ejection fraction [1][2][3]. The exact absolute risks for HF progression from stage A to stage C have remained stable over the past decade (8.4 per 100 person-years), regardless of the implementation of conventional strategies [4]. Previous observational studies found that one-year mortality in HF varied widely from 4% to 45% depending on the presence of acute or chronic HF, HF phenotype, age, gender and concomitant diseases [4][5][6]. According to the Acute Heart Failure Database (AHEAD) registry, liver function test abnormalities (elevations in total bilirubin, γ-glutamyltransferase, alkaline phosphatase, aspartate aminotransferase and alanine aminotransferase) were found in 76% of patients with known acute HF [7]. The PROTECT trial (Placebo-controlled Randomized study of the selective A1 adenosine receptor antagonist KW-3902 for patients hospitalized with acute HF and volume Overload to assess Treatment Effect on Congestion and renal funcTion) and the ASCEND-HF trial (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) showed that acute HF including cardiogenic shock was a meaningful cause of cardiac liver dysfunction [8][9]. Moreover, patients with acute or actually decompensated HF were significantly more likely to have abnormalities in liver test than patients with chronic HF [9]. On the other hand, new-onset liver injury due to cardiac dysfunction has been shown to be a strong predictor for HF progression, higher risks of hospitalization and poor clinical outcomes (all-cause mortality and cardiovascular death) and quality of life [9][10][11]. Along with it, there is evidence of the fact that a severity of liver injury secondary to progressive HF was loosely associated with adverse cardiac remodeling, skeletal muscle dysfunction and sarcopenia/cardiac cachexia [12][13].
However, in HF, multiple pathophysiological interactions between neurohumoral and systemic/local inflammatory activations, as well as an altered immune response, lead not only to adverse cardiac remodeling but also to worsening hepatic circulation and sequelae following the development of acute cardiogenic liver injury and congestive hepatopathy [14][15][16]. The pathogenesis of cardiac hepatopathy is complex and involves underlying canonical molecular mechanisms that overlap with the development and progression of HF (i.e., centrilubular liver necrosis, dilated sinusoids and perisinusoidal fibrosis due to hypoperfusion associated with fluid retention and passive congestion, altered electrolyte and protein metabolism, iron homeostasis and secondary portal hypertension). Additionally, other pathophysiological pathways link liver dysfunction to renal dysfunction, altered endothelial function, anabolic/catabolic imbalance, abnormalities in the intestinal microbiome, impaired metabolism of adipose tissue and skeletal muscle [17][18][19].
2. Definition, Morphological Criteria, and Biochemical Profiling of Liver Damage in HF
The current paradigm of cardiac hepatopathy refers to any hepatocyte injury caused by acute or chronic cardiac dysfunction in the absence of clear evidence of other possible causes of liver injury [20][21]. Although there is no consensus on the definition and terminology of cardiac hepatopathy, the majority of experts use the terms congestive cardiac hepatopathy (CCH) and acute cardiogenic liver injury (ACLI) to describe two different aspects of the disease [22][23][24][25]. CCH is the most common disease in HF caused by passive venous congestion of the liver. Several chronic cardiac diseases, such as constrictive pericarditis, tricuspid regurgitation, primary and secondary cardiomyopathies, inherited cardiac defects and cardiac hypertrophy, associated with chronic HF, usually lead to congestion and thus to the development of CCH [26][27]. There is ample clinical evidence that CCH is closely related to HFrEF/HFmrEF and corresponds to the New York Heart Association (NYHA) HF functional class [28]. In contrast, ACLI is most commonly associated with arterial hypoperfusion and downstream hypoxia due to acute HF resulting from acute myocardial infarction, acute decompensated chronic HF, progressive natural history of severe myocarditis and cardiomyopathies [29]. Nevertheless, the role of primary hypotension in the development of ACLI is controversial. For example, a retrospective analysis of a cohort of 31 HF patients with clinical and biochemical evidence of ACLI revealed that hypotension alone cannot be considered as a trigger of acute liver injury [30]. Rather, other factors such as multiple concomitant diseases in collaboration with various epigenetic influences are implicated for the occurrence of ACLI [31].
Histologically, these two phenotypes of cardiac hepatopathy appear sufficiently distinct. In most cases, CCH is characterized by sinusoidal dilatation associated with extravasation of red blood cells from sinusoids into periportal areas and replacement of hepatocytes by red blood cells, as well as the expression of necrotic areas without severe cellular infiltration and apoptosis of cells in the third zone of Rappaport acini [20][32]. To note, there is evidence of the fact that the small diameter of fenestrae in sinusoids in humans is likely to be a serious obstacle for hepatocyte transduction, so that higher variability of clinical presentation of CCH due to passive congestion may relate to genetic reasons [32]. The extension of periportal necrotic zones together with secondary centrilobular necrosis and accumulation of fibrosis tissue may eventually lead to ineffective intrahepatic circulation supporting ischemia/hypoxia and loss of functional hepatocytes, endothelialization of sinusoids and development of liver cirrhosis in advanced cases [20][33]. The main histological findings in ACLI are massive necrosis of the third zone of Rappaport acini, gross deformation of the liver parenchyma, large fibrotic areas along with rapidly progressive portal hypertension and splenic hypertrophy, often leading to the early onset of hepato-renal syndrome [20][34].
In this context, the primary laboratory findings of cardiac hepatopathy vary depending on numerous factors that include not only the phenotype of the disease, but also the duration of hypotension, the type of cardiac dysfunction (left-sided, right-sided, or biventricular), the presence of comorbidities, patient age and gender [35][36]. Indeed, the greatest increase in transaminases was observed in patients with right-sided or biventricular HF than in left-sided HF [36]. Meanwhile, the prevalence of CCD in HF patients depended on a signature of comorbidity, male gender and older age [7][8][9]. Thus, cardiac hepatopathy may manifest as asymptomatic elevation of serum levels of aminotransferases and/or bilirubin and severe liver injury with significant elevation of aminotransferases, alkaline phosphatase, γ-glutamyltranspeptidase, lactate dehydrogenase and a decrease in plasma albumin levels. Clinical signs of cholestasis, ascites, peripheral edema, portal hypertension and concomitant oligoanuria may not be closely related to the phenotype of cardiac hepatopathy [37]. However, regardless of its phenotype, cardiac hepatopathy has strong prognostic value in identifying all-cause mortality, cardiovascular events and mortality associated with HF and has some particular implications for the management of patients undergoing cardiac assist device implantation or heart transplantation [38].
3. Common Underlying Molecular Mechanisms of Cardiac Hepatopathy
Figure 1 illustrates the main underlying molecular mechanisms of the development of both phenotypes of cardiac hepatopathy. Primary liver ischemia/reperfusion and/or passive liver congestion with secondary tissue ischemia/hypoxia together with oxidative stress are thought to trigger a secondary locally and systemically inflammatory cascade that induces microvascular impairment, intrahepatic thrombosis, liver necrosis/apoptosis, and development of liver fibrosis with its conversion to cirrhosis and interorgan interactions during HF pathogenesis. Along with it, there is assumption of a causative role of sinusoidal thrombi, which are direct reason for tissue ischemia and fibrosis. In addition, alterations in liver function lead to progressive changes in splanchnic blood flow, coronary circulation and systemic hemodynamics and thereby maintain liver fibrosis progression. Finally, cardiac hepatopathy is supported by inadequate secretion of various hepatokines, such as adropin, fetuin-A, FGF-21, alpha-1-microglobulin and selenoprotein P, which influence energy metabolism, local and systemic inflammation and immune response.
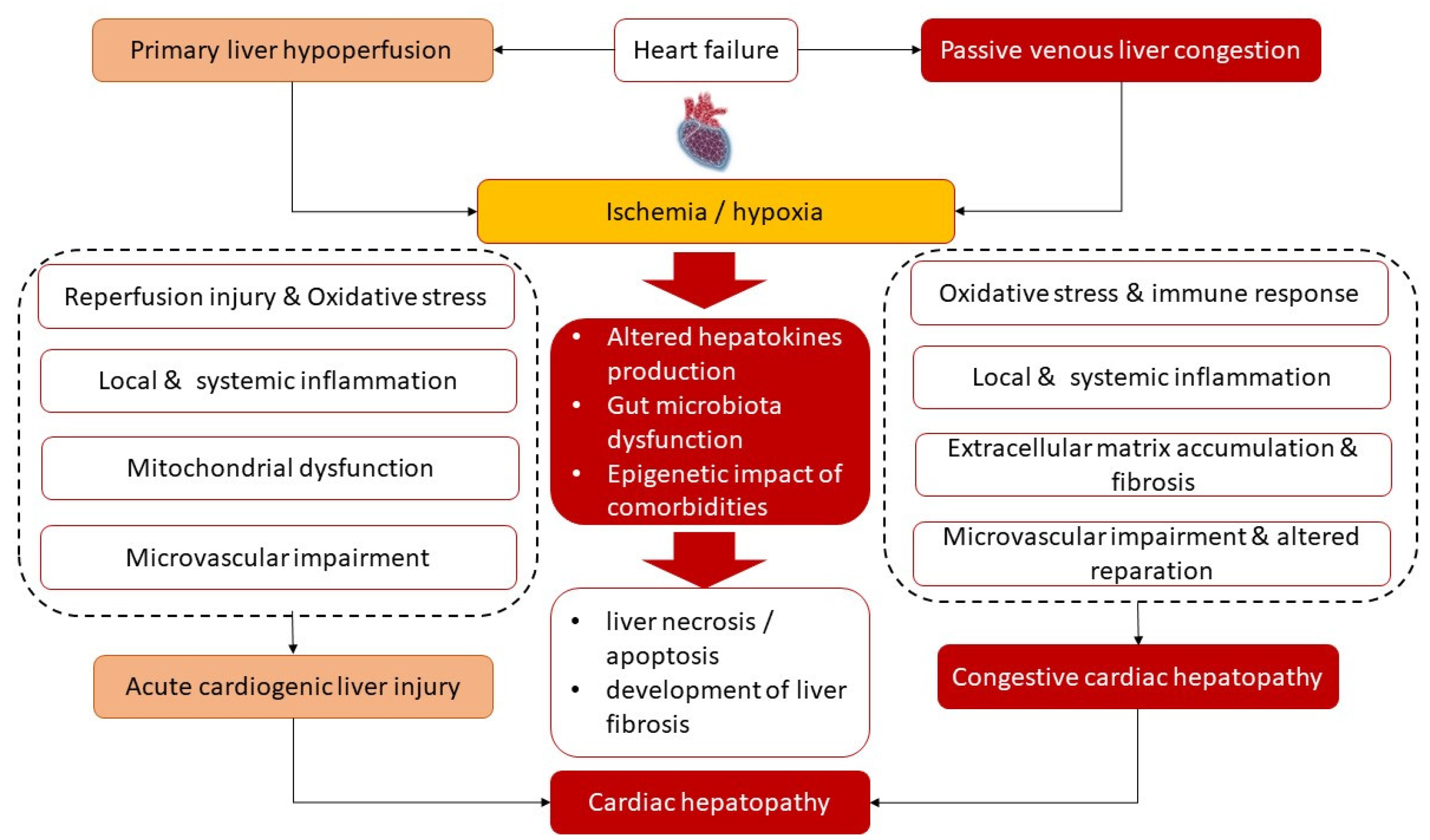
Figure 1. Most common underlying mechanisms of the pathogenesis of cardiac hepatopathy.
3.1. Ischemia/Reperfusion and Inflammation/Fibrosis Cascade
Ischemic injury of the liver is a component of ACLI induced by hypoxia due to hypoperfusion and is characterized by sequential damage to intracellular organelles and whole cells, cell swelling and further persistent disruption of Na+/K+-ATPase function [39][40]. The intracellular accumulation of low-oxidized lipids and proteins, overexpression of xanthine oxidase and NADPH oxidase leads to overproduction of reactive oxygen (ROS) and nitrogen species (such as peroxynitrite, hypochlorite), contributing to acidosis-induced suppression of mitochondrial transmembrane malate-aspartate exchange and carnitine-related mechanism of acyl-CoA transfer, and inducing ischemic mitochondrial dysfunction leading to alteration of mitochondrial permeability and ATP depletion [41]. In fact, the additional damage to liver tissue is a consequence of the paradoxically exacerbating restoration of perfusion by oxygen/Ca2+ delivery [42]. Finally, ROS, intracellular calcium overload, inflammatory cytokines (interleukin [IL]-1b, IL-2, tumor necrosis factor-alpha [TNF]) and chemokines (hypoxia-inducible factor-1α, C-X-C motif ligand-8, C-C motif ligand-2, C-C motif ligand-10) support the early activation of Kupffer cells, the late activation of polymorphic mononuclear cells and the accumulation of CD4+ T lymphocytes. Additionally, these processes activate stellate cells, mononuclear cells and platelets in perisinusoidal spaces and periportal areas and the post-ischemic disruption of liver microcirculation together with a decrease in sinusoidal density in liver parenchyma [43][44]. Antigen-presenting cells and CD4+ T lymphocytes secrete a variety of growth factors such as TNF-β, granulocyte-macrophage colony-stimulating factor and interferon gamma, which enable direct activation of Kupffer cells and promote their ability to synthesize and release inflammatory cytokines [45]. Meanwhile, in ACLI, nitric oxide levels were found to be sufficiently reduced [46]. Moreover, an imbalance between nitric oxide production by nitric oxide synthase and endothelin-1 leads to vasoconstriction of sinusoids and exacerbates the vicious cycle of altered blood circulation [47]. Overall, ischemic and post-ischemic oxidative stress and mitochondrial injury are considered potent triggers for further overexpression of inflammatory genes and activation of hepatocellular apoptosis, ferroptosis and necrosis during hepatic ischemia/reperfusion injury in acute HF [48]. Indeed, Tanaka et al. (2014) [48] found that numerous apoptotic hepatocytes in the third zone of Rappaport acinar were co-localized with NADPH oxidase 4 (NOX4) and that these findings were a consequence of hypoxia-induced intrahepatic microcirculatory failure but were not induced by activated APCs, such as macrophages and mononuclear cells. On the other hand, hepatocyte energy metabolism, as measured by determining the local activities of citrate synthase, carnitine palmitoyltransferase-1 and cytochrome c oxidase, was increased by inflammatory cytokines despite ultrastructural changes in mitochondria during hepatocyte apoptosis [49]. It is possible that the transmission of death signals in the early phase of hepatocyte apoptosis is mainly associated with the co-stimulation of APCs, while in the late phase it is regulated by other signals such as NOX4 and active caspase-3, Bax and Bcl-2 that might be regulated via Toll-like receptor-4 (TLR4)/phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/glycogen synthase kinase 3-beta (GSK-3β) signaling [50]. However, local production of inflammatory cytokines mediates expression of cell surface adhesion molecules (intracellular cell adhesion molecule, vascular cell adhesion molecule) on the surfaces of hepatocytes and endothelial cells, induces adherence of APCs and consequently leads to intravascular/intra-sinusoidal coagulation, which contributes to microcirculatory failure and autophagy [51][52]. Interestingly, ischemia/reperfusion-induced liver injury may be promoted by M1 polarization of macrophages via regulation of peroxisome proliferator-activated receptor-γ (PPAR-γ) in response to increasing anaerobic glycolysis and accumulation of lactic acid in the microenvironment [53]. Evidence suggests that polarization of hepatic M1 macrophages, which supports acute and chronic liver injury, can be promoted by gut-derived exosomes following intestinal ischemia/reperfusion [52][53]. Thus, decreased splanchnic blood flow is an independent factor contributing to liver injury. However, all these triggers prolong hepatic ischemia/hypoxia and exacerbate apoptosis/necrosis, creating a vicious cycle of excessive inflammatory response involving activated antigen presenting cells with excessive cytokine and ROS production, leading to further oxidative liver tissue damage [54][55].
The molecular pathways contributing to CCH are not different from those mentioned above, while ischemia is secondary to passive liver congestion and the reperfusion phase is closely related to decompensation of HF associated with unstable hemodynamics. Indeed, CCH is not as dramatic compared with ACLI, so the histological sequelae are not necessarily the same as those of ACLI. Initial dilatation of hepatic sinusoids due to passive liver congestion is associated with a degree of venous pressure in vena porta and results in the exudation of red blood cells, activated mononuclear cells, platelets and protein rich-fluid into the perisinusoidal space of Disse. However, low-grade inflammation and excessive fibrosis play a more significant role in the pathogenesis of CCH than in ACLI. There is strong evidence that inflammatory chemokine signaling (C-X-C motif ligand-8, C-C motif ligand-2, C-C motif ligand-10) derived from activated Kupffer cells induces polarization of monocyte-derived macrophages into the M1 phenotype and stimulates fibrogenesis through activation of hepatic stellate cells [56][57][58]. Another difference between CCH and ACLI concerns the advocacy of hepatic angiogenesis [59]. Indeed, hypoxia-induced vascular endothelial growth factor (VEGF) expression has been found to be crucial in advanced fibrosis, whereas inflammation in early stages of fibrotic transformation of the liver may involve VEGF in hepatic angiogenesis [59][60].
3.2. Intestinal Microbiota
Recent data show that the secretome of the gut microbiota may be involved in the regulation of liver regeneration after acute and chronic liver injury [61]. Therefore, different microbiota profiles were found to be associated with the rate of decompensation HF and thereby intervened in outcome in these patients [62]. Indeed, a wide range of secretory components, including gut-derived lipopolysaccharides, bile acids associated with gut microbiota and numerous bacterial metabolites (short-chain fatty acids and tryptophan metabolites), may influence protective capacity by protecting hepatocytes from injury and supporting repair [63]. Although there is no certain explanation for the transmission of signals from the microbiota to hepatocytes [64], the secretome of the intestinal microbiota is thought to mediate activities of the farnesoid X receptor (FXR)-fibroblast growth factor 19 (FGF19) axis and enhance the TGR5-glucagon-like peptide axis, which are involved in the metabolic regulation of proliferative response and attenuation of immunological imbalance [65][66]. In addition, lipopolysaccharide (LPS) derived from bacterial walls of the intestinal microbiota and bacterial DNAs, that are known TLR9 agonists, have been found at elevated levels in the peripheral blood of patients with various liver diseases [67][68]. Increased permeability of the intestinal vascular barrier to such macromolecules is the result of sufficient disruption of splanchnic blood flow, edema of the intestinal wall, free fluid in the peritoneum and maldigestion [69]. Finally, simultaneous stimulation of antigen-presenting cells by LPS and bacterial DNAs may lead to activation of the inducible form of nitric oxide synthase and release of nitric oxide, which is accompanied by systemic vasodilation and hyperdynamic changes in the circulation to prevent impaired perfusion of distant tissues [68][70][71]. Yet, several metabolites of the microbiota, such as trimethylamine N-oxide, tryptamine and indole-3-acetate, may attenuate inflammatory responses, insulin resistance, mitochondrial and endoplasmic reticulum dysfunction through binding with endoplasmic reticulum stress kinase PERK (EIF2AK3) and activation of transcription factor FoxO1, as well as they may mediate the expression of fatty acid synthase and sterol regulatory element-binding protein-1c in hepatocytes [72][73]. Normally, these metabolites reduced fatty-acid- and LPS-stimulated ability of macrophages to synthetize and release pro-inflammatory cytokines and inhibited the migration activity of T cells and mononuclears toward a chemokine [74]. Depletion of the microbiota in HF was associated with liver fibrosis due to LPS-stimulation of stellate cells and, however, promoting local iron sequestration through ferroportin-expressing phagocytes and hepatocyte-derived hepcidin acting as activator of conventional dendritic cells [74]. To note, about 30% of HF patients had no changes in the microbiota, while bacterial translocation from the gut microbiota and persistence of bacterial antigens were assumed to be responsible for systemic inflammation and HF decompensation. These facts indicate that the gut microbiota may influence liver architectonic and perfusion through other mechanisms, which need to be discovered [62].
3.3. Adipose Tissue Dysfunction
Adipose tissue is involved in various changes in HF patients (inflammation, browning) and it is a source of mesenchymal stem cells and adipokines, which essentially regulate the energy metabolism of distant tissues including the liver [75][76][77]. Adipogenic liver transformation propagates local redox imbalance and activates in lipid-dependent deterioration of hepatocytes. Alterations in hepatocyte histology is associated with elevated expression of PPARγ, adipocyte protein, interleukin-6 (IL-6), interleukin-18 (IL-18), CD36 and adiponectin [78]. It appears that PPARα and PPARδ dysfunction is a key regulator, through which adipokines can mediate repair processes in the liver [79]. Nevertheless, altered autophagy/mitophagy and mitochondrial dysfunction may be attenuated by adiponectin in both ACLI and CCH [80]. It is possible that adiponectin induces autophagosome formation through AMP-activated protein kinase (AMPK)-dependent activation of Unc-51-like kinase 1, which subsequently leads to the removal of damaged mitochondria from hepatocytes [80]. Indeed, suppression of autophagy in white adipose tissues attenuated the liver fibrosis through adipose-liver crosstalk [81]. However, adiponectin protects liver injury by inactivating the TGF-beta-1/SMAD2 pathway and exerts an anti-fibrotic effect via the AMPK/STAT3 pathway [82]. However, adipokines (adiponectin, leptin) are thought to contribute to the anti-inflammatory effects of various hepatokines, such as apelin, adropin and fetuin-A by decreasing the local production of IL-6 and fibroblast growth factor 21 (FGF21), which prevent hepatic steatosis and fibrosis [83][84][85][86][87]. At the same time, apelin mediats Fas-induced liver injury in part via activation of c-Jun N-terminal kinases [88]. In parallel, FOXO transcription factors, which are directly modulated by insulin signaling in the liver, can be downregulated by IL-6, TNF-alpha and follistatin and can be upregulated by FGF21 [89]. Thus, adipokines that modulate the synthesis of IL-6, TNF-alpha, FGF21/Klotho and nitric oxide production via extracellular regulated kinase1/2 (p-ERK1/2) may improve liver metabolism by indirectly regulating glucose homeostasis and preventing mitochondrial dysfunction [90][91][92]. Meanwhile, FGF21 acts on adipocytes and renal cells to induce synthesis of angiotensin-converting enzyme 2 that inhibits hypertension and reverses vascular damage and microvascular inflammation [87]. Overall, the functional pleiotropism of adipose tissue affects its ability to synthesize and secretes numerous adipokines, which regulate liver metabolism, thermogenesis, microvascular inflammation, liver and remote tissue reparations, angiogenesis, insulin resistance, endothelial integrity, and causes not only multiple serious conditions, including adverse cardiac remodeling, adipose tissue dysfunction, liver fibrosis, but also mediates adaptive local changes in target organs.
3.4. Impaired Skeletal Muscles Metabolism
Skeletal muscle myopathy is a common condition of HF, especially in patients with HFrEF and HFmrEF, which is strongly associated with a decrease in effective perfusion [93]. However, skeletal muscle cells have endocrine capabilities and secrete a broad spectrum of regulatory proteins called myokines (myonectine, irisin, myoststin, FGF21, FGF15), which are involved in autocrine/paracrine regulation of distant organs and tissues, including the liver [94]. They are able to activate AMP-activated protein kinase and modulate its cooperation with the transcription factor nuclear respiratory factor 1, sirtuin-1 and the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α, and induce the expression of mitochondrial transcription factor-A genes that support mitochondrial protection [95][96][97]. Loss of a pool of these secreting proteins such as irisin, myonectin, FGF21 and growth differential factor-11 (GDF11) reduces the tissue-protective effect of myokines [98][99][100]. However, a precise understanding of the beneficial effects of myokines on cardiac hepatopathy appears to require further investigation.
3.5. Epigenetic Impact of Metabolic Comorbidities on Liver Tissue Modification
Epigenetic changes in DNA through DNA methylation, histone modifications and microRNA sequences are thought to be critical for specific changes in genes that coordinate several underlying molecular mechanisms in the pathogenesis of cardiac hepatopathy [101]. In addition, various metabolic comorbidities such as diabetes mellitus, insulin resistance and nonalcoholic fatty liver disease are thought to influence the reparative capacity of the liver through epigenetic changes in genes encoding adipokines, transport proteins (microsomal triglyceride transfer protein, patatin-like phospholipase domain-containing protein 3), PPAR-γ receptors and also genes involved in adipogenic/lipogenic regulation (homeostasis-associated gene, retinoid X receptor alpha gene, and liver X receptor alpha gene) [102][103][104][105]. However, epigenetic regulation of liver tissue susceptibility to acute and chronic damage in HF is the subject of further investigation.
Thus, the occurrence of cardiac hepatopathy is the result of several overlapping mechanisms, which include ischemia/reperfusion injury of liver tissue, passive fluid congestion, reduced hepatic blood with intrahepatic thrombosis, total body hypoxemia, and inability to utilize oxygen and metabolic components. These mechanisms are under close regulation of auto-paracrine and epigenetic influences. Increasing evidence suggests that interplay between hepatokines, adipokines and cardiokines mainly natriuretic peptides are crucial for the metabolic crosstalk between systemic and local myocardial/microvascular inflammation in HF and peripheral tissue damage in liver and some remote organs and tissues including kidney, spleen, skeletal muscle and adipose, tissue), but the direct underlying mechanisms linking advanced cardiac dysfunction and liver fibrosis remain not fully elucidated.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12020516
References
- Emmons-Bell, S.; Johnson, C.; Roth, G. Prevalence, incidence and survival of heart failure: A systematic review. Heart 2022, 108, 1351–1360.
- Van Riet, E.E.; Hoes, A.W.; Wagenaar, K.P.; Limburg, A.; Landman, M.A.; Rutten, F.H. Epidemiology of heart failure: The prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur. J. Heart Fail. 2016, 18, 242–252.
- Cvijic, M.; Rib, Y.; Danojevic, S.; Radulescu, C.I.; Nazghaidze, N.; Vardas, P. Heart failure with mildly reduced ejection fraction: From diagnosis to treatment. Gaps and dilemmas in current clinical practice. Heart Fail. Rev. 2022.
- Echouffo-Tcheugui, J.B.; Erqou, S.; Butler, J.; Yancy, C.W.; Fonarow, G.C. Assessing the Risk of Progression from Asymptomatic Left Ventricular Dysfunction to Overt Heart Failure: A Systematic Overview and Meta-Analysis. JACC Heart Fail. 2016, 4, 237–248.
- Gori, M.; Redfield, M.M.; Calabrese, A.; Canova, P.; Cioffi, G.; De Maria, R.; Grosu, A.; Fontana, A.; Iacovoni, A.; Ferrari, P.; et al. Is mild asymptomatic left ventricular systolic dysfunction always predictive of adverse events in high-risk populations? Insights from the DAVID-Berg study. Eur. J. Heart Fail. 2018, 20, 1540–1548.
- Pandhi, J.; Gottdiener, J.S.; Bartz, T.M.; Kop, W.J.; Mehra, M.R. Comparison of characteristics and outcomes of asymptomatic versus symptomatic left ventricular dysfunction in subjects 65 years old or older (from the Cardiovascular Health Study). Am. J. Cardiol. 2011, 107, 1667–1674.
- Vyskocilova, K.; Spinarova, L.; Spinar, J.; Mikusova, T.; Vitovec, J.; Malek, J.; Malek, F.; Linhart, A.; Fedorco, M.; Widimsky, P.; et al. Prevalence and clinical significance of liver function abnormalities in patients with acute heart failure. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech Repub. 2015, 159, 429–436.
- Biegus, J.; Hillege, H.L.; Postmus, D.; Valente, M.A.; Bloomfield, D.M.; Cleland, J.G.; Cotter, G.; Davison, B.A.; Dittrich, H.C.; Fiuzat, M.; et al. Abnormal liver function tests in acute heart failure: Relationship with clinical characteristics and outcome in the PROTECT study. Eur. J. Heart Fail. 2016, 18, 830–839.
- Samsky, M.D.; Dunning, A.; DeVore, A.D.; Schulte, P.J.; Starling, R.C.; Tang, W.H.; Armstrong, P.W.; Ezekowitz, J.A.; Butler, J.; McMurray, J.J.; et al. Liver function tests in patients with acute heart failure and associated outcomes: Insights from ASCEND-HF. Eur. J. Heart Fail. 2016, 18, 424–432.
- Ambrosy, A.P.; Vaduganathan, M.; Huffman, M.D.; Khan, S.; Kwasny, M.J.; Fought, A.J.; Maggioni, A.P.; Swedberg, K.; Konstam, M.A.; Zannad, F.; et al. Clinical course and predictive value of liver function tests in patients hospitalized for worsening heart failure with reduced ejection fraction: An analysis of the EVEREST trial. Eur. J. Heart Fail. 2012, 14, 302–311.
- Wang, H.Y.; Huang, Y.; Chen, X.Z.; Zhang, Z.L.; Gui, C. Prognostic potential of liver injury in patients with dilated cardiomyopathy: A retrospective study. Eur. J. Med. Res. 2022, 27, 237.
- Ambrosy, A.P.; Vaduganathan, M.; Mentz, R.J.; Greene, S.J.; Subačius, H.; Konstam, M.A.; Maggioni, A.P.; Swedberg, K.; Gheorghiade, M. Clinical profile and prognostic value of low systolic blood pressure in patients hospitalized for heart failure with reduced ejection fraction: Insights from the Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study with Tolvaptan (EVEREST) trial. Am. Heart J. 2013, 165, 216–225.
- Meng, W.; Wang, L.; Fan, H.; Mao, S.; Song, X.; Zhang, Z. Total Bilirubin Level is Associated with the Risk of Left Atrial Appendage Thrombosis in Patients with Non-Valvular Atrial Fibrillation. Glob. Heart 2022, 17, 90.
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart Failure and Liver Disease: Cardiohepatic Interactions. JACC Heart Fail. 2019, 7, 87–97.
- Laribi, S.; Mebazaa, A. Cardiohepatic syndrome: Liver injury in decompensated heart failure. Curr. Heart Fail. Rep. 2014, 11, 236–240.
- Branchereau, M.; Burcelin, R.; Heymes, C. The gut microbiome and heart failure: A better gut for a better heart. Rev. Endocr. Metab. Disord. 2019, 20, 407–414.
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells 2020, 9, 567.
- Gheorghiade, M.; Follath, F.; Ponikowski, P.; Barsuk, J.H.; Blair, J.E.; Cleland, J.G.; Dickstein, K.; Drazner, M.H.; Fonarow, G.C.; Jaarsma, T.; et al. Assessing and grading congestion in acute heart failure: A scientific statement from the acute heart failure committee of the heart failure association of the European Society of Cardiology and endorsed by the European Society of Intensive Care Medicine. Eur. J. Heart Fail. 2010, 12, 423–433.
- Çağlı, K.; Başar, F.N.; Tok, D.; Turak, O.; Başar, Ö. How to interpret liver function tests in heart failure patients? Turk. J. Gastroenterol. 2015, 26, 197–203.
- De Gonzalez, A.K.K.; Lefkowitch, J.H. Heart Disease and the Liver: Pathologic Evaluation. Gastroenterol. Clin. North Am. 2017, 46, 421–435.
- Fouad, Y.M.; Yehia, R. Hepato-cardiac disorders. World J. Hepatol. 2014, 6, 41–54.
- Hakuno, D.; Kimura, M.; Ito, S.; Satoh, J.; Nakashima, Y.; Horie, T.; Kuwabara, Y.; Nishiga, M.; Ide, Y.; Baba, O.; et al. Hepatokine α1-Microglobulin Signaling Exacerbates Inflammation and Disturbs Fibrotic Repair in Mouse Myocardial Infarction. Sci. Rep. 2018, 8, 16749.
- Kavoliuniene, A.; Vaitiekiene, A.; Cesnaite, G. Congestive hepatopathy and hypoxic hepatitis in heart failure: A cardiologist’s point of view. Int. J. Cardiol. 2013, 166, 554–558.
- Birrer, R.; Takuda, Y.; Takara, T. Hypoxic hepatopathy: Pathophysiology and prognosis. Intern. Med. 2007, 46, 1063–1070.
- Mauriello, J.N.; Straughan, M.M. Right-Sided Heart Failure and the Liver. Crit. Care Nurs. Clin. North Am. 2022, 34, 341–350.
- Correale, M.; Tarantino, N.; Petrucci, R.; Tricarico, L.; Laonigro, I.; Di Biase, M.; Brunetti, N.D. Liver disease and heart failure: Back and forth. Eur. J. Intern. Med. 2018, 48, 25–34.
- Megalla, S.; Holtzman, D.; Aronow, W.S.; Nazari, R.; Korenfeld, S.; Schwarcz, A.; Goldberg, Y.; Spevack, D.M. Predictors of cardiac hepatopathy in patients with right heart failure. Med. Sci. Monit. 2011, 17, CR537–CR541.
- Allen, L.A.; Felker, G.M.; Pocock, S.; McMurray, J.J.; Pfeffer, M.A.; Swedberg, K.; CHARM Investigators. Liver function abnormalities and outcome in patients with chronic heart failure: Data from the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) program. Eur. J. Heart Fail. 2009, 11, 170–177.
- Benincasa, G.; Cuomo, O.; Vasco, M.; Vennarecci, G.; Canonico, R.; Della Mura, N.; Alfano, R.; Napoli, C. Epigenetic-sensitive challenges of cardiohepatic interactions: Clinical and therapeutic implications in heart failure patients. Eur. J. Gastroenterol. Hepatol. 2021, 33, 1247–1253.
- Seeto, R.K.; Fenn, B.; Rockey, D.C. Ischemic hepatitis: Clinical presentation and pathogenesis. Am. J. Med. 2000, 109, 109–113.
- Lightsey, J.M.; Rockey, D.C. Current concepts in ischemic hepatitis. Curr. Opin. Gastroenterol. 2017, 33, 158–163.
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; De Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene. Ther. 2008, 15, 1193–1199.
- Sundaram, V.; Fang, J.C. Gastrointestinal and Liver Issues in Heart Failure. Circulation 2016, 133, 1696–1703.
- Goncalvesova, E.; Kovacova, M. Heart failure affects liver morphology and function. What are the clinical implications? Bratisl. Lek. Listy. 2018, 119, 98–102.
- Moreira-Silva, S.; Urbano, J.; Moura, M.C.; Ferreira-Coimbra, J.; Bettencourt, P.; Pimenta, J. Liver cytolysis in acute heart failure: What does it mean? Clinical profile and outcomes of a prospective hospital cohort. Int. J. Cardiol. 2016, 221, 422–427.
- Møller, S.; Dümcke, C.W.; Krag, A. The heart and the liver. Expert. Rev. Gastroenterol. Hepatol. 2009, 3, 51–64.
- Samsky, M.D.; Patel, C.B.; DeWald, T.A.; Smith, A.D.; Felker, G.M.; Rogers, J.G.; Hernandez, A.F. Cardiohepatic interactions in heart failure: An overview and clinical implications. J. Am. Coll. Cardiol. 2013, 61, 2397–2405.
- Scalzo, N.; Canastar, M.; Lebovics, E. Part 2: Disease of the Heart and Liver: A Relationship That Cuts Both Ways. Cardiol. Rev. 2022, 30, 161–166.
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G15–G26.
- Lee, W.Y.; Lee, J.S.; Lee, S.M. Protective effects of combined ischemic preconditioning and ascorbic acid on mitochondrial injury in hepatic ischemia/reperfusion. J. Surg. Res. 2007, 142, 45–52.
- Hasegawa, T.; Malle, E.; Farhood, A.; Jaeschke, H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: Attenuation by ischemic preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G760–G767.
- Jin, L.M.; Liu, Y.X.; Zhou, L.; Xie, H.Y.; Feng, X.W.; Li, H.; Zheng, S.S. Ischemic preconditioning attenuates morphological and biochemical changes in hepatic ischemia/reperfusion in rats. Pathobiology 2010, 77, 136–146.
- Mendes-Braz, M.; Elias-Miró, M.; Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Ramalho, F.S.; Peralta, C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J. Biomed. Biotechnol. 2012, 2012, 298657.
- Sun, C.K.; Zhang, X.Y.; Zimmermann, A.; Davis, G.; Wheatley, A.M. Effect of ischemia-reperfusion injury on the microcirculation of the steatotic liver of the Zucker rat. Transplantation 2001, 72, 1625–1631.
- Nastos, C.; Kalimeris, K.; Papoutsidakis, N.; Tasoulis, M.K.; Lykoudis, P.M.; Theodoraki, K.; Nastou, D.; Smyrniotis, V.; Arkadopoulos, N. Global consequences of liver ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2014, 2014, 906965.
- Uhlmann, D.; Glasser, S.; Gaebel, G.; Armann, B.; Ludwig, S.; Tannapfel, A.; Hauss, J.; Witzigmann, H. Improvement of postischemic hepatic microcirculation after endothelin A receptor blockade—Endothelin antagonism influences platelet-endothelium interactions. J. Gastrointest. Surg. 2005, 9, 187–197.
- Kuwano, A.; Kurokawa, M.; Kohjima, M.; Imoto, K.; Tashiro, S.; Suzuki, H.; Tanaka, M.; Okada, S.; Kato, M.; Ogawa, Y.; et al. Microcirculatory disturbance in acute liver injury. Exp. Ther. Med. 2021, 21, 596.
- Tanaka, M.; Tanaka, K.; Masaki, Y.; Miyazaki, M.; Kato, M.; Kotoh, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Intrahepatic microcirculatory disorder, parenchymal hypoxia and NOX4 upregulation result in zonal differences in hepatocyte apoptosis following lipopolysaccharide- and D-galactosamine-induced acute liver failure in rats. Int. J. Mol. Med. 2014, 33, 254–262.
- Zhang, X.; Jiang, W.; Zhou, A.L.; Zhao, M.; Jiang, D.R. Inhibitory effect of oxymatrine on hepatocyte apoptosis via TLR4/PI3K/Akt/GSK-3β signaling pathway. World J. Gastroenterol. 2017, 23, 3839–3849.
- Quesnelle, K.M.; Bystrom, P.V.; Toledo-Pereyra, L.H. Molecular responses to ischemia and reperfusion in the liver. Arch. Toxicol. 2015, 89, 651–657.
- Wang, X.T.; Tang, Y.B.; Lin, Q.Q.; Wang, X.Y.; Song, Z.Y.; Hao, M.L.; Qian, W.; Wang, W.T. Role of autophagy in liver injury induced by lung ischemia/reperfusion in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2022, 38, 102–107. (In Chinese)
- Zhao, J.; Chen, X.D.; Yan, Z.Z.; Huang, W.F.; Liu, K.X.; Li, C. Gut-Derived Exosomes Induce Liver Injury After Intestinal Ischemia/Reperfusion by Promoting Hepatic Macrophage Polarization. Inflammation 2022, 45, 2325–2338.
- Ding, W.; Duan, Y.; Qu, Z.; Feng, J.; Zhang, R.; Li, X.; Sun, D.; Zhang, X.; Lu, Y. Acidic Microenvironment Aggravates the Severity of Hepatic Ischemia/Reperfusion Injury by Modulating M1-Polarization Through Regulating PPAR-γ Signal. Front. Immunol. 2021, 12, 697362.
- Chen, L.Y.; Yang, B.; Zhou, L.; Ren, F.; Duan, Z.P.; Ma, Y.J. Promotion of mitochondrial energy metabolism during hepatocyte apoptosis in a rat model of acute liver failure. Mol. Med. Rep. 2015, 12, 5035–5041.
- Teodoro, J.S.; Da Silva, R.T.; Machado, I.F.; Panisello-Roselló, A.; Roselló-Catafau, J.; Rolo, A.P.; Palmeira, C.M. Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria. Cells 2022, 11, 688.
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096.
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072.
- Sun, Y.Y.; Li, X.F.; Meng, X.M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2017, 85, 166–174.
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Baeck, C.; Hittatiya, K.; Eulberg, D.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971.
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 371–390.
- Zheng, Z.; Wang, B. The Gut-Liver Axis in Health and Disease: The Role of Gut Microbiota-Derived Signals in Liver Injury and Regeneration. Front. Immunol. 2021, 12, 775526.
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The microbiota in cirrhosis and its role in hepatic decompensation. J. Hepatol. 2021, 75 (Suppl. 1), S67–S81.
- Liu, H.X.; Keane, R.; Sheng, L.; Wan, Y.J. Implications of microbiota and bile acid in liver injury and regeneration. J. Hepatol. 2015, 63, 1502–1510.
- Baker, S.S.; Baker, R.D. Gut Microbiota and Liver Injury (II): Chronic Liver Injury. Adv. Exp. Med. Biol. 2020, 1238, 39–54.
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A story of liver and gut microbes: How does the intestinal flora affect liver disease? A review of the literature. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G889–G906.
- Behary, J.; Amorim, N.; Jiang, X.T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187.
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169.
- Bellot, P.; García-Pagán, J.C.; Francés, R.; Abraldes, J.G.; Navasa, M.; Pérez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052.
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834.
- La Villa, G.; Gentilini, P. Hemodynamic alterations in liver cirrhosis. Mol. Aspects Med. 2008, 29, 112–118.
- Chu, C.J.; Lee, F.Y.; Wang, S.S.; Chang, F.Y.; Lin, H.C.; Lu, R.H.; Chan, C.C.; Lee, S.D. Splanchnic endotoxin levels in cirrhotic rats induced by carbon tetrachloride. Zhonghua Yi Xue Za Zhi 2000, 63, 196–204.
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111.
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151.e5.
- Bessman, N.J.; Mathieu, J.R.R.; Renassia, C.; Zhou, L.; Fung, T.C.; Fernandez, K.C.; Austin, C.; Moeller, J.B.; Zumerle, S.; Louis, S.; et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 2020, 368, 186–189.
- Teshima, T.; Matsumoto, H.; Michishita, M.; Matsuoka, A.; Shiba, M.; Nagashima, T.; Koyama, H. Allogenic Adipose Tissue-Derived Mesenchymal Stem Cells Ameliorate Acute Hepatic Injury in Dogs. Stem Cells Int. 2017, 2017, 3892514.
- Kim, M.D.; Kim, S.S.; Cha, H.Y.; Jang, S.H.; Chang, D.Y.; Kim, W.; Suh-Kim, H.; Lee, J.H. Therapeutic effect of hepatocyte growth factor-secreting mesenchymal stem cells in a rat model of liver fibrosis. Exp. Mol. Med. 2014, 46, e110.
- Berezin, A.E.; Berezin, A.A. Impaired function of fibroblast growth factor 23/Klotho protein axis in prediabetes and diabetes mellitus: Promising predictor of cardiovascular risk. Diabetes Metab. Syndr. 2019, 13, 2549–2556.
- Lakhani, H.V.; Sharma, D.; Dodrill, M.W.; Nawab, A.; Sharma, N.; Cottrill, C.L.; Shapiro, J.I.; Sodhi, K. Phenotypic Alteration of Hepatocytes in Non-Alcoholic Fatty Liver Disease. Int. J. Med. Sci. 2018, 15, 1591–1599.
- Li, T.H.; Yang, Y.Y.; Huang, C.C.; Liu, C.W.; Tsai, H.C.; Lin, M.W.; Tsai, C.Y.; Huang, S.F.; Wang, Y.W.; Lee, T.Y.; et al. Elafibranor interrupts adipose dysfunction-mediated gut and liver injury in mice with alcoholic steatohepatitis. Clin. Sci. 2019, 133, 531–544.
- Lin, Z.; Wu, F.; Lin, S.; Pan, X.; Jin, L.; Lu, T.; Shi, L.; Wang, Y.; Xu, A.; Li, X. Adiponectin protects against acetaminophen-induced mitochondrial dysfunction and acute liver injury by promoting autophagy in mice. J. Hepatol. 2014, 61, 825–831.
- Sakane, S.; Hikita, H.; Shirai, K.; Myojin, Y.; Sasaki, Y.; Kudo, S.; Fukumoto, K.; Mizutani, N.; Tahata, Y.; Makino, Y.; et al. White Adipose Tissue Autophagy and Adipose-Liver Crosstalk Exacerbate Nonalcoholic Fatty Liver Disease in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1683–1699.
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445.
- Clemens, M.M.; Kennon-McGill, S.; Vazquez, J.H.; Stephens, O.W.; Peterson, E.A.; Johann, D.J.; Allard, F.D.; Yee, E.U.; McCullough, S.S.; James, L.P.; et al. Exogenous phosphatidic acid reduces acetaminophen-induced liver injury in mice by activating hepatic interleukin-6 signaling through inter-organ crosstalk. Acta Pharm. Sin. B 2021, 11, 3836–3846.
- Tilg, H.; Kaser, A.; Moschen, A.R. How to modulate inflammatory cytokines in liver diseases. Liver Int. 2006, 26, 1029–1039.
- Wolf, A.M.; Wolf, D.; Avila, M.A.; Moschen, A.R.; Berasain, C.; Enrich, B.; Rumpold, H.; Tilg, H. Up-regulation of the anti-inflammatory adipokine adiponectin in acute liver failure in mice. J. Hepatol. 2006, 44, 537–543.
- Serbetçi, K.; Uysal, O.; Erkasap, N.; Köken, T.; Baydemir, C.; Erkasap, S. Anti-apoptotic and antioxidant effect of leptin on CCl₄-induced acute liver injury in rats. Mol. Biol. Rep. 2012, 39, 1173–1180.
- Li, F.; Chen, J.; Liu, Y.; Gu, Z.; Jiang, M.; Zhang, L.; Chen, S.Y.; Deng, Z.; McClain, C.J.; Feng, W. Deficiency of Cathelicidin Attenuates High-Fat Diet Plus Alcohol-Induced Liver Injury through FGF21/Adiponectin Regulation. Cells 2021, 10, 3333.
- Yasuzaki, H.; Yoshida, S.; Hashimoto, T.; Shibata, W.; Inamori, M.; Toya, Y.; Tamura, K.; Maeda, S.; Umemura, S. Involvement of the apelin receptor APJ in Fas-induced liver injury. Liver Int. 2013, 33, 118–126.
- Garcia Whitlock, A.E.; Sostre-Colón, J.; Gavin, M.; Martin, N.D.; Baur, J.A.; Sims, C.A.; Titchenell, P.M. Loss of FOXO transcription factors in the liver mitigates stress-induced hyperglycemia. Mol. Metab. 2021, 51, 101246.
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794.
- Deng, Z.H., Jr.; Yan, G.T.; Wang, L.H.; Zhang, J.Y.; Xue, H.; Zhang, K. Leptin relieves intestinal ischemia/reperfusion injury by promoting ERK1/2 phosphorylation and the NO signaling pathway. J. Trauma Acute Care Surg. 2012, 72, 143–149.
- Ikejima, K.; Honda, H.; Yoshikawa, M.; Hirose, M.; Kitamura, T.; Takei, Y.; Sato, N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology 2001, 34, 288–297.
- Berezin, A.E.; Berezin, A.A.; Lichtenauer, M. Myokines and Heart Failure: Challenging Role in Adverse Cardiac Remodeling, Myopathy, and Clinical Outcomes. Dis. Markers 2021, 2021, 6644631.
- Pang, B.P.S.; Chan, W.S.; Chan, C.B. Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle-Do Myokines Play a Role? Antioxidants 2021, 10, 179.
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109.
- Pan, X.; Shao, Y.; Wu, F.; Wang, Y.; Xiong, R.; Zheng, J.; Tian, H.; Wang, B.; Wang, Y.; Zhang, Y.; et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1-7) Axis in Mice. Cell Metab. 2018, 27, 1323–1337.e5.
- Nagatomo, I.; Nakanishi, K.; Yamamoto, R.; Ide, S.; Ishibashi, C.; Moriyama, T.; Yamauchi-Takihara, K. Soluble angiotensin-converting enzyme 2 association with lipid metabolism. Front. Med. 2022, 9, 955928.
- Landecho, M.F.; Tuero, C.; Valentí, V.; Bilbao, I.; de la Higuera, M.; Frühbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664.
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265.
- Musso, G.; Gambino, R.; De Michieli, F.; Durazzo, M.; Pagano, G.; Cassader, M. Adiponectin gene polymorphisms modulate acute adiponectin response to dietary fat: Possible pathogenetic role in NASH. Hepatology 2008, 47, 1167–1177.
- Gambino, R.; Cassader, M.; Pagano, G.; Durazzo, M.; Musso, G. Polymorphism in microsomal triglyceride transfer protein: A link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology 2007, 45, 1097–1107.
- Stasinou, E.; Argyraki, M.; Sotiriadou, F.; Lambropoulos, A.; Fotoulaki, M. Association between rs738409 and rs2896019 single-nucleotide polymorphisms of phospholipase domain-containing protein 3 and susceptibility to nonalcoholic fatty liver disease in Greek children and adolescents. Ann. Gastroenterol. 2022, 35, 297–306.
- Zhang, R.N.; Shen, F.; Pan, Q.; Cao, H.X.; Chen, G.Y.; Fan, J.G. PPARGC1A rs8192678 G>A polymorphism affects the severity of hepatic histological features and nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease. World J. Gastroenterol. 2021, 27, 3863–3876.
This entry is offline, you can click here to edit this entry!