1. Cellular Origin of CAFs in HCC
With the understanding that CAFs have many subtypes, it was thought that experimental and clinical studies would provide insights into the nature and extent of CAF heterogeneity and the biological characteristics of CAFs [
9]. The development of novel coculture models and implementation of single-cell RNA sequencing (scRNA-seq) techniques have provided detailed information on the origins of CAFs and revealed the presence of high levels of CAF heterogeneity in many cancer types [
11,
18]. Furthermore, novel genetically engineered mouse models (GEMMs) have substantially advanced our information about fibroblast origins, heterogeneity, plasticity and functions. Currently, there are no lineage-tracing models for CAFs [
18]. However, intravital-microscopy imaging techniques associated with GEMMs could evaluate the origins and plasticity of activated CAF subtypes [
9,
17,
18]. Experimental studies that utilized lecithin retinol acyltransferase-cyclization recombination enzyme (Lrat-Cre) and PDGF receptor β (PDGFRβ)-Cre have reported that hepatic stellate cells (HSCs) are the dominant source of myofibroblasts in liver cirrhosis [
10,
18]. Under physiological conditions, HSCs are quiescent and reside in the space of Disse. Following liver injury, HSCs lose their retinyl esters in lipid droplets, become activated and transdifferentiate into myofibroblasts that produce ECM components and αSMA [
12,
32]. TGFβ plays a key role in development and progression of liver cirrhosis and HCC. During hepatocarcinogenesis, TGFβ promotes transdifferentiation of HSCs into CAFs [
12,
42]. Another study documented that hepatocyte-derived PDGF-C could also transdifferentiate HCCs into myofibroblasts to foster progression of HCC [
10,
42]
Figure 3.
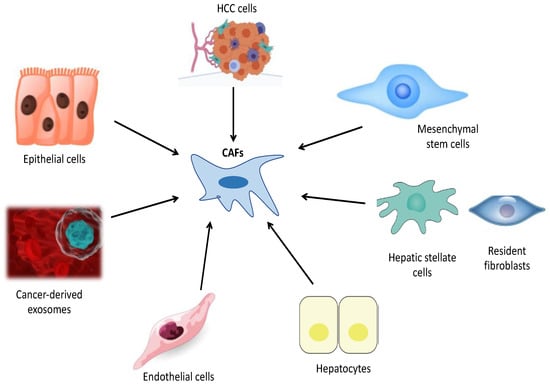
Figure 3. This figure indicates potential cells from which cancer-associated fibroblasts (CAFs) originate in HCC: HCC cells, Mesenchymal stem cells, hepatic stellate cells, resident fibroblasts, hepatocytes, endothelial cells, cancer-derived exosomes and epithelial cells.
Although the molecular basis of the cancer cell has been studied extensively, the mechanisms that activate CAFs and regulate their recruitment are only beginning to be elucidated. Many studies have demonstrated that the genetic characteristics of cancer cells play a critical role in formation of TMEs. As such, CAF signatures detected in different cancers can be utilized for stratification purposes and enable prognostic information about clinical outcomes [
18,
42]. Several factors have been shown to promote activation and reprogramming of CAFs, including epithelial cues such as IL-1, PDGF, metabolic reprogramming, oxidative stress, stromal cues, microRNAs and epigenetic alterations [
18]. Implementation of novel genetic fate mapping and scRNA-seq techniques has enabled more favorable evidence in monitoring the origins of CAFs [
10,
15,
16,
42]. scRNA-seq provides high-resolution pictures of the transcriptomes of single cells. It is now widely used to identify cell populations within specific tissues, including that of the liver. Recently, a study that analyzed scRNA-seq data from mouse-liver cells indicated that transcription factor 21 (Tcf21) is a specific marker that distinguishes quiescent HSCs from other liver-cell types and activated HSCs [
43]. Tracing the fate of Tcf21-CreER
+ cells under normal conditions as well as after various chronic liver injuries, that study found that Tcf21-CrER
+ preferentially marked periportal and pericentral HSCs that were quiescent in the steady state but became activated in the DEN/CCL
4-induced state, originating 85% of the CAFs in HCC [
43]. Another important finding of this study was that Tcf21-CreER
+-targeted perivenous stellate cells are the main source of myofibroblasts and CAFs in chronically injured livers [
43]. More recently, another study that utilized genetic tracing in combination with scRNA-seq analysis, as well as genetic depletion through Cr-lox-mediated deletion approaches, demonstrated that CAFs are derived primarily from resident HSCs. A second key finding of this study was that interactions between HSC-derived CAFs and tumor cells account for a main mechanism of tumor promotion and restriction in desmoplastic liver cancer, and the majority of CAFs express HSC signatures abundantly [
44].
Mesenchymal stem cells (MSCs) are multipotent cells capable of differentiating into various cell types, including adipocytes, cartilage, bone and other cells, in physiological conditions [
19,
45,
46,
47,
48]. MSCs have been thought to have great potential for liver regeneration and a therapeutic effect on liver fibrosis [
48,
49].
In addition to their high regenerative capacities, MSCs can be recruited within the HCC TME and exhibit significant roles in modulation of HCC progression, growth, metastasis and drug sensitivity [
47,
48]. They have tumor-promoting and tumor-restricting functions in HCC; however, the mechanisms that underlie these opposing effects are not fully understood [
48]. In HCC patients, MSCs were demonstrated to promote apoptosis and inhibit HCC-cell proliferation, migration and invasion. Tumor-infiltrating MSCs within the HCC TME can transdifferentiate into CAF-like phenotypes after being acclimated via cancer cells [
46,
47]. A novel trial that investigated the impact of the HCC TME on human-adipose MSCs (hA-MSCs) and predicted hA MSC intracellular miRNA’s role demonstrated that when cocultured with Huh7 cells, the MSCs substantially upregulated the expressions of CAF markers, including αSMA, vimentin, c-MYC, MMP2, VEGF and IL-6, and thus, the hA-MSCs could transdifferentiate into CAF-like phenotypes [
48]. The second key finding of that study was crosstalk between the HCC cells and components of the HCC TME to generate these CAFs [
48]. Another study, which addressed the effects of the TME on differentiation of MSCs into CAFs, demonstrated that after exposure to epithelial hepatic carcinoma SK-Hep1 cells, MSCs may acquire the molecular and functional characteristics of CAFs [
49]. However, the implementations of only in vitro cell-line cocultivation assays in both studies is their major limitation, and in vivo lineage-tracing experiments are required to determine transdifferentiation of MSCs into CAFs in the future [
10].
Emerging evidence suggests that cancer-derived exosomes, which play a key role in carcinogenesis and tumor-cell proliferation, transdifferentiate into CAFs through a novel mechanism of endothelial-to-mesenchymal transition (EndMT) [
32,
50]. Epithelial cells, through EMTs or endothelial cells via EndMT, can acquire mesenchymal cell characteristics, which can be another source of CAFs [
10]. Greening et al. have reported that cancer-derived exosomes induced EndMT through promotion of proliferation of endothelial cells and reconstituted premetastatic niches that formed a TME in a metastatic region [
51]. They also revealed that exosome-deficient cancer cells abolish fibroblast differentiation and inhibit tumor-cell growth through silencing of exosome-secretion regulator Rab27b [
51]. Cancer-derived exosomes are thought to be mediators that regulate interactions between stromal cells and reshape TMEs [
51]. In another study, MSC-derived exosomes were found to inhibit EndMT, promote angiogenesis and maintain vascular homeostasis, while cancer-derived exosomes triggered EndMT, followed by induction of CAFs [
50].
Multiple studies have reported that TGFβ1, a profibrotic growth factor, promotes adult hepatocytes to undergo phenotypic and functional properties of EMTs [
10,
12,
17,
18]. In lineage-tracing experiments that utilized AlbCre;R26Rstoplac Z double transgenic mice, researchers demonstrated that a substantial population (up to 45%) of FSP1-positive fibroblasts was derived from hepatocytes through EMTs [
52]. Similar results were obtained in kidney studies that reported that approximately 40% of all fibroblasts originate via EMTs. However, this finding may be controversial; another experimental trial, which used triple-transgenic mice that expressed ROSA26 stop β-galactosidase, albumin Cre and collagen α1 green fluorescent protein (GFP), demonstrated that type-1 collagen-producing cells do not originate from hepatocytes and that hepatocytes in vivo neither acquire mesenchymal marker expression nor exhibit myofibroblast-like morphology [
53].
In some specific conditions, HCC cells may undergo EMTs and express markers of CAFs [
10,
12,
18]. For example, fibroblast activation protein (FAP) expression has been reported in some cancer cells as well as in CAFs, which correlates with poor clinical consequences [
16,
54]. FAPs can be induced under hypoxia, which is also crucial in the biological behavior of cancer cells [
10,
16,
20,
47]. Recent studies that investigated the expression levels of FAPs and hypoxia inducible factor 1α (HIF-1α) in HCC cells demonstrated that hypoxia can induce upregulation of FAPs in HCC cells and be indicative of poor prognosis in patients with HCC [
10,
54]. HIF-1α promotes tumor cells to acquire booster proliferation, invasion and metastasis capabilities under the metabolic stress conditions in which HIF-1α degradation is inhibited [
16]. Xu et al. have reported that CAF-derived CCL5 promotes HCC metastasis through the HIF-1α/ZEB1 pathway [
16]. Furthermore, they demonstrated that CCL5 was positively correlated with HIF1-α in clinical samples, and high levels of expression of HIF1-α were associated with worse overall survival [
16]. CAFs secrete TGFβ, which exhibits both protumor and antitumor functions through diverse mechanisms [
55]. Similar to TGFβ, nuclear liver X receptors (LXRs) either suppress or promote cancer through inhibition of cell proliferation or assistance of tumor cells in avoidance of immune surveillance [
56,
57]. A recent study reported that a majority of epithelial HCC cells expressed detectable LXRα levels and responded to LXR agonists, and that LXRs limit TGFβ-dependent CAF differentiation [
58]. Another study, which utilized the in vitro EndMT model, documented the transdifferentiation of fetal-liver sinusoidal endothelial cells into fibroblast-like cells while mesenchymal markers were increased and the endothelial markers were decreased [
59]. However, an important limitation was that the functional properties of CAFs were not investigated in these studies.
2. Impact of CAFs on HCC Progression
The number of studies of CAFs has increased dramatically in the last decade, with recognition that CAFs are the most significant component of the stromal cell population in a TME. CAFs are often implicated in HCC progression and drug resistance [
60,
61]. They modulate HCC progression through various mechanisms, including direct effects on HCC cells through secretion of soluble factors and exosomes and indirect effects through other stromal cells and ECM remodeling [
21,
23]. Recently, based on scRNA-seq data from the TCGA and GEO databases, Yu et al. identified four CAF subpopulations in HCC, three of which have been associated with prognosis in patients with HCC [
62]. Of the total four hundred and twenty-three analyzed genes, six were primarily linked with 39 pathways, such as those of angiogenesis, apoptosis and hypoxia. Another significant finding of this trial was that risk signatures were substantially associated with stromal and immune scores, as well as some immune cells. In another study, Qi et al. showed that CAF-derived exosomal miR-20a-5p facilitated HCC progression through the LIM domain and actin binding 1 (LIMA1)-mediated β-catenin pathway [
63]
Table 1.
Previous studies have revealed that CAFs promote cancer progression and metastasis through production of a variety of soluble factors, including inflammatory cytokines, growth factors and chemokines [
12,
16,
19,
20,
21,
22,
23,
64]. However, CAFs are phenotypically and functionally heterogeneous and can exhibit both protumorigenic and antitumorigenic activity [
65,
66,
67,
68]. A recent trial that implemented proteomics and sc-RNS-seq analysis in order to examine the CAF landscape in HCC demonstrated three major CAF populations in HCC, one of which specifically expresses the prolargin protein that binds and inhibits activity of several proangiogenic proteins, including hepatocyte and fibroblast growth factors. As such, prolargin is thought to be an angiogenesis modulator and a CAF-derived tumor suppressor in HCC [
68]. Studies that primarily investigated the Hedgehog (Hh) signaling pathway indicated that CAFs could also have antitumoral activity in some conditions [
12,
16,
17,
18]. CAFs with specific proteomic profiles can exhibit a tumor-inhibitory function. A trial that addressed stromal transcriptional signatures in some tumors showed the presences of various stromal transcriptional signatures in tumor tissue samples, and aggressive tumors were associated with distinct stromal signatures [
15,
16,
17,
18]. More recently, Song et al. reported detailed cytokine-regulated crosstalk between CAFs, HCC cells and TANs, fostering tumor-cell migration and invasion [
69]
Figure 4.
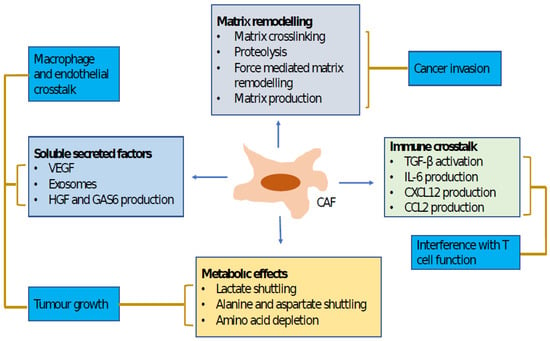
Figure 4. This figure indicates cancer-associated fibroblast functions and the mechanisms that regulate them. Lines connect mechanisms to functions. Both matrix remodeling and production of soluble factors promote tumor-cell invasion. Soluble factors also play a critical role in tumor-cell growth and changes in tumor microenvironments, which are also influenced by the altered metabolic states of tumors. CAF, cancer-associated fibroblast; CCL2, CC-chemokine ligand 2; CXCL12, CXC-chemokine ligand 12; IL-6, interleukin 6; GAS6, growth arrest-specific protein 6; HGF, hepatocyte growth factor; TGFβ, transforming growth factor-β; VEGF, vascular endothelial growth factor.
In 2011, Mazzocca et al. demonstrated, for the first time, the existence of crosstalk between CAFs and HCC cells [
70]. They indicated the molecular and functional differences between peritumoral fibroblasts (PTFs) and CAFs [
70,
71]. Additionally, they showed that HCC-derived lysophostatidic acid (LPA) plays a critical role in promotion of transdifferentiation of PTFs to CAF-like myofibroblastic phenotypes, which in turn accelerates HCC progression [
70]. Similar to stem cells, tumor-initiating cells are orchestrated through various signals generated within their TMEs. Lau et al. revealed that CAF-derived hepatocyte growth factor (HGF) orchestrates tumor-initiating cell plasticity in HCC through activation of c-Met/FRA1/HEY1 signaling [
30]. Furthermore, they found that HGF-induced FRA1 activation was associated with the fibrosis-dependent development of HCC in a STAM NASH-HCC mouse model [
30]. Another significant finding of this study was that the presence of αSMA-positive CAFs correlated with worse clinical consequences [
30]. Another trial, which used an in vitro model of paracrine interaction between HCC-cell lines (HepG2, SNU423) and HSC and investigated the regulatory mechanism that underlies keratin 19 (KRT19) expression in HCC, demonstrated that KRT 19 expression in HCC is orchestrated through fibroblast-derived hepatocyte growth factor (HGF) via a MET-ERK1/2-AP1 and SP1 signaling pathway [
72].
Previous studies have shown that CAFs are closely related to invasion and metastasis of HCC cells, but the mechanisms of CAFs that drive HCC metastasis were not completely clarified [
73,
74]. CAFs do not exist independently around tumors, but crosstalk with tumor cells to promote their malignant phenotypes [
73,
74]. Tumor cells can recruit CAF precursors and transdifferentiate normal fibroblasts into CAFs. Meanwhile, CAFs secrete large amounts of cytokines, chemokines, growth factors and ECM proteins, which form TMEs to promote HCC-cell proliferation, metastasis and drug resistance [
16,
75,
76]. The chemokine–chemokine receptor (CK-CKR) network represents a key regulator of immune-cell recruitment and shapes the TME [
77]. Studies have indicated that CAFs upregulate levels of CCL2, CCL5, CCL7, CCL26 and CXCL17 and acquire a booster-tumor metastatic phenotype [
23]. CCL7 and CXCL16 promote both migration and invasion of HCC cells, while CCL2 and CCL5 promote only migration of HCC cells [
78]. Moreover, CCL2 and CCL5 activate the Hh signaling pathway, while CCL7 and CXCL16 boost the activity of TGFβ in HCC cells. More recently, Xu et al. found that CCL5 was the most significant cytokine in the CAFs that promote HCC metastasis; they also observed that serum CCL5 levels were quite high in patients who developed HCC in cirrhotic livers [
16]. CCL5, an inflammatory cytokine, plays a relevant role in CAF promotion of carcinogenesis [
79]. Hypoxia-inducible factor 1 alpha (HIF1α) exhibits crucial roles in regulation of energy metabolism, angiogenesis and other processes in the TME and provides cancer cells to gain more proliferation and metastasis capacity. It has been reported that CAFs have been involved in regulation of tumor HIF1α to promote tumor progression [
16,
17,
18,
42]. Xu et al. indicated that CAF-derived CCL5 inhibited HIF1α ubiquitination degradation, maintained HIF1α expression under normoxia and promoted an EMT and metastasis through activation of downstream factor ZEB1 [
16]. Meanwhile, CCL5 was positively correlated with HIF1α in clinical samples; these high expressions were significantly associated with poor prognosis [
16].
Recently, CAFs have been documented to recruit immune cells within the TME, including neutrophils and macrophages [
80,
81]. Endosialin is a transmembrane protein that is expressed in some cancer cells, stromal cells and pericytes, but barely expressed in normal tissue. In a novel study, Yang et al. documented that endosialin is particularly expressed in CAFs in HCC and its expression is associated with worse overall survival in HCC patients [
82]. They also observed that endosialin could regulate expression of growth arrest-specific protein 6 (GAS6] in CAFs, which promotes M2 polarization of macrophages to promote HCC progression [
82]. Additionally, another trial that investigated the impact of HCC-derived CAFs on neutrophils revealed that the HCC-derived CAFs induced chemotaxis in the neutrophils and protected them from spontaneous apoptosis [
83]. These researchers found that the HCC-derived CAFs promoted activation of STAT3 pathways in the neutrophils, which was essential for the survival and function of the activated neutrophils [
83]. One of the significant findings of this study was that HCC-derived CAFs primed neutrophils’ impaired T-cell function through the PD1/PDL1 signaling pathway [
83]. It has been suggested that HCC-derived CAFs regulate survival, activation and function of neutrophils within HCC through an IL6-STAT3-PDL1 signaling pathway, which represents a novel mechanism for the role of CAFs in remodeling the cancer niche and provides a potential target for HCC therapy [
15].
Table 1. Impact of CAFs on HCC progression.
Although interaction between HCC cells, CAFs and other stromal cells in the HCC TME has been well documented, the nature of the complex interaction between CAFs and other components within the TME has not yet been completely elucidated, mainly during the distinct HCC stage [
71]. Recently, Song et al. demonstrated complex interactions between HCC cells, CAFs and TANs, which enhance cancer stemness and recruitment of TANs in HCC [
69]. They reported, for the first time, that CAFs isolated from advanced-stage HCC showed a greater tumor-promoting effect in vivo than those isolated from the early stages [
20]. This study highlighted the clinical significance of CAF-derived cardiotrophin-like cytokine factor 1 (CLCF1) signaling in CAF-mediated direct crosstalk with tumor cells and indirect interaction with TANs within the HCC TME [
69]. Furthermore, the researchers thereof reported that CAF-derived CLCF1 upregulated two key cytokines, chemokine (C-X-C motif) ligand 6 (CXCL6) and TGFβ, in HCC cells through the Akt/extracellular signal-regulated kinase 1/2 (ERK1/2)/STAT3 signaling pathway, which in turn induced HCC-cell stemness and TAN infiltration and polarization [
69]. Clinically, high levels of CLCF1 expression have been found to be correlated with aggressive tumor behaviors and worse clinical outcomes [
69]. In HCC, tumor cells secrete a large amount of TGFβ, while CAFs produce relatively lower levels of TGFβ, IL-6 and granulocyte-colony-stimulating factor (GCSF) to promote N2 neutrophils [
15,
69,
71]. These data clearly indicate that CAFs predominantly induce HCC progression via paracrine pathways [
69,
71]. However, other mechanisms through which CAFs orchestrate HCC progression, including exosomes and extracellular vesicles, can play a role in this progression [
15,
17,
18,
69,
71]. CLCF1-promoted CXCL6 and TGFβ constitute the crucial bridge that connects cellular crosstalk between CAFs, HCC cells and TANs [
69]. The study thereof reported that CXCL6 fosters HCC stemness via transcriptional driving of E2F1 [
69,
71]. Additionally, microRNA (miRNA) that is involved in E2F1 dysregulation exhibits a critical role in tumor progression, and CXCL6 influences miRNA function in carcinogenesis [
69,
71].
Neutrophils are innate immune cells that are thought to be a double-edged sword during carcinogenesis [
15,
83,
84,
85,
86,
87,
88]. Distinct TMEs polarize neutrophils to antitumorigenic (N1) or protumorigenic (N2) phenotypes [
15,
83,
84]. N2 TANs have the capacity to form neutrophil extracellular traps (NETs), which can act to promote HCC development in the setting of cirrhosis [
15,
84]. Emerging evidence reveals that high infiltration levels of tumor-associated neutrophils (TANs) within the TME are correlated with worse overall survival in some solid tumors [
15]. TANs promote cancer progression through induction of tumor-cell proliferation, metastasis and stemness; remodeling of the ECM; augmentation of angiogenesis; or stimulation of immunosuppression. In HCC [
15,
69,
85,
88,
89,
90,
91], TANs have been reported to increase tumor-cell stemness and recruit immunosuppressive macrophages and Tregs [
88]. However, their exact role in hepatocarcinogenesis and the effects of TMEs on education about TANs, regarding their phenotypes and functions, are largely unknown. Multiple studies have highlighted the relevance of interaction between HCC cells, TANs and CAFs in affecting HCC progression [
15,
17,
18,
69]. CAFs can suppress neutrophil function through the SDF1a/CXCR4/IL-6 pathway, which promotes production of CD66b, PD-L1, CXCL8/IL-8, TNF and CCL2, which can inhibit the function and proliferation of T cells in vitro [
82]. CAF-derived CLCF1 can promote tumoral expression of CXCL6 and TGFβ, resulting in neutrophil recruitment and N2 polarization, respectively [
69]. TANs acquired a protumoral N2 phenotype in the middle and advanced stages in correlation with increased levels of CLCF1 [
69]. In advanced HCC, high levels of CLCF1 expression result in an augmented CLCF1-CXCL6/TGFβ pathway, recruiting more TANs and polarizing them toward the N2 phenotype to further facilitate tumor progression [
15,
69,
71]. As such, CLCF1 may be a potential prognostic biomarker for HCC, and selective blocking of CLCF1 signaling could provide an effective therapy for HCC patients [
15].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24043941