Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Heme proteins (or hemeproteins or hemoproteins) are a structurally and functionally diverse group of metalloproteins exhibiting the heme moiety—an iron-coordinated porphyrin ring—as the prosthetic group. NO is biosynthesized endogenously by heme proteins named NO synthases (NOSs), which oxidize the guanidino group of L-arginine into L-citrulline and NO in the presence of O2 and reduced nicotinamide-adenine-dinucleotide phosphate (NADPH).
- nitric oxide
- heme
- S-nitrosylation
- NO
1. Reactivity of NO with the Heme Group
Recent reviews on heme-binding proteins extensively cover the complex reactivity of NO with the metal center of the heme group [24,25]. For several heme proteins, the interaction or the reactivity with NO is widely accepted as physiologically relevant, whereas for others—although intrinsically reactive with NO in vitro—the relevance is under debate. For circulating proteins such as Hb, several reactions might be relevant in vivo at different times, depending on the local microenvironment they encounter within the bloodstream. The main reactivities that heme proteins exhibit are reported below and in Figure 1a.
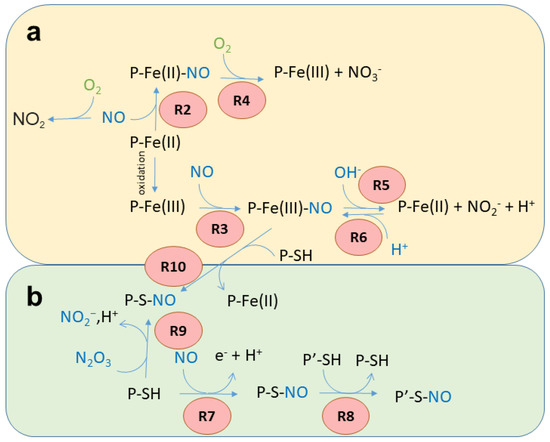
Figure 1. Possible reactions of NO with the heme group (a) and Cys residues (b). The reaction numbers (red circles) refer to the reactions reported in the text.
Reversible binding to ferrous heme. Under anaerobic conditions, Fe(II) heme proteins rapidly bind NO to produce a stable nitrosyl complex (Reaction 2, R2 in Figure 1a). For instance, Hb exhibits a kNO of 60 µM−1s−1 [26]. The binding is reversible, but the dissociation rate constants are typically very small, with koff < 10-3 s−1 [27,28].
P-Fe(II) + NO → P-Fe(II)-NO
In vivo, this reaction is associated with NO scavenging only under anaerobic conditions, since the presence of heme-bound O2 would quickly result in NO dioxygenation (see Reaction 4).
Reversible binding to ferric heme. The Fe(III) heme group forms bonds 6–7 orders of magnitude weaker with NO in comparison with Fe(II) (Reaction 3, R3 in Figure 1a) [24,25]. For Mb, the kNO for this reaction is 0.070 µM−1s−1 [29,30], with koff typically >10 s−1.
P-Fe(III) + NO → P-Fe(III)-NO
Some heme proteins play a physiological role based on this reaction. For instance, NPs are heme-binding lipocalins found in the saliva of the bloodsucking Reduviidae and Cimicidae families of Heteroptera. They are stable in the Fe(III) state, and the structure of the distal side of the heme group stabilizes the NO complex and prevents reactions with O2, water, hydroxides, or thiols, thus allowing their function as transporters of NO [31]. NO is then released at the feeding site, where it induces vasodilation.
NO dioxygenation. In the presence of O2, several Fe(II) heme proteins, and particularly globins, are known to catalyze the NO-dioxygenase (NOD) reaction, where NO is oxidized to nitrate (NO3−) via a peroxynitrite intermediate (Reaction 4, R4 in Figure 1a):
P-Fe(II)- O2 + NO → P-Fe(III) N(O)OO → P-Fe(III) + NO3−
This reaction, which results in heme oxidation, is very fast (Hb kNOD 60–80 µM−1s−1 [26]) when the heme group is bound to O2 and exposed to NO. When the heme protein is bound to NO and exposed to O2, the reaction occurs at a much slower rate, since NO has to dissociate to allow O2 binding before the reaction can take place. The NOD reaction in vivo is typically associated with NO scavenging under aerobic conditions [26], but also, as in the case of Ascaris Hb, to O2 scavenging under anaerobic conditions [32]. It has been noted that catalytic reactions that lead to heme oxidation can be physiologically relevant only if efficient mechanisms for its reduction are in place to restore the Fe(II) form [33].
Autoreduction in Fe(III)–NO complexes. Fe(III) heme proteins undergo ‘reductive nitrosylation’ (or ‘autoreduction’) in the presence of NO (Reaction 5, R5 in Figure 1a).
P-Fe(III) + NO + OH− → P-Fe(II)-NO2H → P-Fe(II) + NO2− + H+
The reaction occurs in two steps and is pH-dependent, since it usually requires the displacement of the intermediate by hydroxide [34] (Scheme 1). Since the affinity of heme proteins in the Fe(III) form for NO is usually low (see Reaction 3), this reaction requires NO concentrations that afford a significant binding.
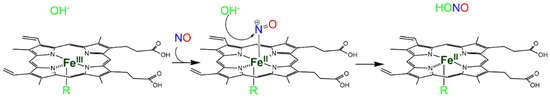
Scheme 1. Hydroxide-mediated reductive nitrosylation of Fe(III) heme b.
Nitrite reduction. The reverse of Reaction 5, i.e., the reduction in NO2− (NiR) by Fe(II) heme in the absence of O2 (Reaction 6, R6 in Figure 3a), has been extensively investigated in mammals because of its potential to produce NO [35].
P-Fe(II) + NO2− + H+→ P-Fe(III) + NO + OH−
At least ten mammalian heme proteins have been characterized in vitro for NiR, i.e., Hb, myoglobin (Mb), Ngb, Cygb, cytochrome c oxidase, cytochrome bc1, cytochrome c, endothelial (eNOS), cytochrome P450, and indoleamine 2,3-dioxygenase 1 [8,16,36,37]. For proteins that are physiologically in the Fe(II) state and exhibit high concentrations (i.e., Mb and Hb in muscle tissue and red blood cells, respectively), the extent of NO production through this reaction is under debate, since newly produced NO would tightly bind the nearby Fe(II) hemes (Reaction 1).
NO2− concentration in mammalian tissues is fairly high (0.1–10 μM [38]), confirming that the NiR reaction can physiologically take place in vivo. NO2− can have a dietary origin [39] or can be produced from nitrate (NO3−) by bacterial Mo-containing nitrate reductases [40], suggesting a NO3−-NO2−-NO pathway for NO biosynthesis that involves the oral microbiome and enteral symbiotic bacteria for the initial step [41,42].
2. S-Nitrosylation
S-nitrosylation is a Cys post-translational modification that reversibly regulates protein function and is a fundamental regulatory mechanism in NO-related signaling [22,43]. It consists of the covalent binding of a nitroso group to a Cys residue to form the SNO. S-nitrosylation may produce novel protein–protein interactions by producing changes in the surface-charge or by allosterically generating solvent-exposed binding sites [44]. The current view is that S-nitrosylation plays a role equivalent to phosphorylation or acetylation to elicit the biological responses of target proteins, and the modification is often considered as an evolutionarily conserved signaling mechanism that involves many classes of proteins located in the cardiovascular system [45].
Regardless of the reaction responsible for S-nitrosylation, it is believed that not all exposed Cys residues can form stable SNOs. The identification of S-nitrosylation sites in the proteome [46] has allowed the identification of Cys environments that either promote the formation of SNOs or stabilize them once they are produced. Particularly, the presence of an acid-base motif in the proximity of the reactive Cys was associated with the formation of a thiolate, thus favoring the reaction [47]. Since the environment of Cys side chains can depend on the conformation of the protein within its conformational space, the residue can be reactive in one conformation but not in another. For example, it was proposed that in oxygenated Hb (R-state), the histidine (His) residue proximal to Cysβ93 favors the base-catalyzed S-nitrosylation, whereas, in the deoxygenated form (T-state), denitrosylation is facilitated by the proximal aspartic acid (Asp) residue [47]. In serine racemase, the reaction with either NO or nitroso donors is also conformation-dependent and occurs only in the conformation stabilized by its allosteric effector ATP [48]. Hydrophobic environments, such as biological membranes or environments formed by protein structures, favor the formation of SNOs ([47] and references herein).
In the last two decades, S-nitrosylation has drawn attention mostly for the biomedical implications for its direct effect on a variety of signaling pathways in physiological and pathological conditions [49]. In several neurodegenerative diseases, high levels of NO may contribute to mitochondrial dysfunction and protein misfolding via aberrant protein S-nitrosylation. Aberrant protein S-nitrosylation may particularly impact the quality control of protein folding, including molecular chaperones, autophagy, and the ubiquitin-proteasome system [49].
The main reactions in which Cys residues and NO are involved have been summarized in Figure 1b and detailed below.
S-nitrosylation associated with reduction. It is accepted that the direct reaction of the NO radical with thiols does not result in S-nitrosylation since the reaction requires the loss of an electron. However, in the presence of O2 or other oxidants, a redox reaction can occur, with the formation of the SNO through a thiyl radical or through a nitrosonium (NO+) ion [22] (Reaction 7, R7 in Figure 1b).
P-SH + NO → P-SNO + e− + H+
S-nitrosylation by transnitrosylation. S-nitrosylation can also result from transnitrosylation by S-nitrosoglutathione (GSNO) [43] or S-nitrosylated proteins [50,51] (Reaction 8, R8 in Figure 1b).
P-S-NO+ P′-SH → P-SH + P′SNO
Transnitrosylation has been associated with site-specific protein–protein interactions mediated by specific recognition motifs [47] (and references herein) (Scheme 2). Enzymes that donate and accept nitroso groups from proteins are named S-nitrosylases and denitrosylases, respectively, and can have high-molecular weight final acceptors—as the thioredoxin/thioredoxin reductase system (Trx1/TrxR) —or low molecular weight acceptors, as the GSNO reductase and SNO-coenzyme A reductase systems [52,53]. The enzymatic control of S-nitrosylation is fundamental for the regulation of NO-based signal transduction and is dynamically regulated by the equilibrium between S-nitrosylated proteins and low-molecular-weight SNOs, which may be a cause or consequence of diseases, including asthma, cystic fibrosis, Parkinson’s disease, heart failure, and stroke [47].
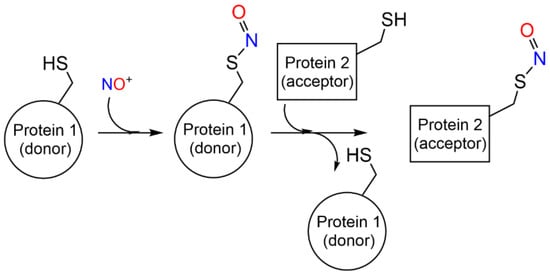
Scheme 2. S-nitrosylation coupled with transnitrosylation of proteins.
S-nitrosylation by reaction with nitrous anhydride. S-nitrosylated Cys residues can also be formed by the reaction with dinitrogen trioxide (or nitrous anhydride, N2O3) (Reaction 9, R9 in Figure 1b). N2O3 partially dissociates, forming the nitrosonium ion (NO+), which reacts with thiolates (Reaction 8, transnitrosylation). N2O3 is formed through the reaction of NO with O2, particularly in hydrophobic microenvironments such as cell membranes [54].
P-S− + N2O3 → P-SNO+ NO2−
Metal-to-Cys S-nitrosylation. Metal-to-Cys S-nitrosylation (or heme-dependent S-nitrosylation or heme-assisted S-nitrosylation in the specific case of heme proteins) is a special case of S-nitrosylation associated with reduction (Reaction 7) and is of great relevance to heme proteins. It consists of the transfer of a nitroso group from a heme group to a nearby Cys residue, either within the same protein (auto S-nitrosylation) or in another protein or peptide (Reaction 10, R10 in Figure 1).
P-Fe(III)-NO+ P-SH → P-SNO + P-Fe(II) + H+
The NO+ ion is generated by the binding of NO to a Fe(III) heme (Reaction 3), which is reduced to Fe(II) (Scheme 3).
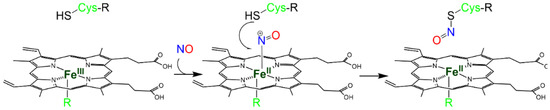
Scheme 3. Metal-to-Cys S-nitrosylation.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12020321
This entry is offline, you can click here to edit this entry!