Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Aging is a complex process characterized by an ongoing decline in physiological functions, leading to degenerative diseases and an increased probability of death. Cellular senescence has been typically considered as an anti-proliferative process; the chronic accumulation of senescent cells contributes to tissue dysfunction and aging. Recognizing the hallmarks of senescence is crucial for the research and development of therapies against aging.
- aging
- biomarker
- senescence
- skin
- hallmark
- SASP
- ROS
1. Introduction
Since the dawn of civilization, humans have been confronted with the problem of aging and mortality and have therefore sought ways to slow this process, if not defeat it altogether (e.g., the philosopher’s stone myth). Currently, the question of delaying aging is as relevant as ever: more than 10% of the world’s population is over 65 years old, and Europe is the “oldest” region, being home to 19% of those over 65 [1]. According to the United Nations forecast report, the total number of people over 60 will double between 2017 and 2050, rising from 962 million to an astounding 2.1 billion [2]. Consequently, our lifespans are rapidly outpacing the so-called health span. Unfortunately, wisdom is not the only gift of maturity; it also bears a heightened risk of geriatric conditions such as frailty, impaired mobility, and cognitive deterioration. Elderly individuals are sick for longer on average, and it is not uncommon for them to struggle with multiple chronic issues simultaneously, placing a sizable burden on the medical system.
In general, aging is viewed as a multifactorial process in which the body changes over time, leading to functional impairment and an ever-increasing likelihood of the loss of life. The problem of aging encompasses a web of aspects, from obvious clinical effects to sociopolitical outcomes for the lives of individuals and society as a whole. These aspects are the subject of the study of gerontology [3]. The subfield of gerontology that deals with the study of aging in vitro by analyzing the mechanisms of aging in cultured cells is called cytogerontology [4]. It should be noted that cytogerontological findings cannot be the sole basis for proposing theories due to scientific reductionism. Most models of normal or altered aging in multicellular organisms are reduced to several specific molecular switches in a set of cells (sometimes limited to a single cell line). As a result, most of these models are devoid of the complexity of neural and humoral influences, which makes them quite vulnerable when translating in vitro data to in vivo models.
In 2013, López-Otín et al. [5] described nine now-canonical hallmarks of cellular and molecular aging and grouped them into three categories. The primary hallmarks include developments at the molecular level: telomere attrition, the instability of the genome, epigenetic alterations, and a loss of proteostasis—all of which have the potential to wreak havoc on the molecular level. Antagonistic hallmarks comprise a counteractive response to the embodiment of the primary hallmarks (deregulated nutrient sensing, mitochondrial dysfunction, and cellular senescence). Finally, integrative hallmarks such as stem cell exhaustion and altered cellular communication are the culmination of damage caused by the aforementioned hallmarks, leading to what is known as a damaged phenotype, i.e., functional deterioration.
While the primary hallmarks, as the name implies, are the starting point of damage, they alone do not constitute evidence of cellular aging. The downstream response is, in a sense, a counterbalance that serves to bring the system to homeostasis after initial detrimental effects. Unfortunately, over time, if left to its own devices, this mechanism can lead to additional damage, which, in turn, causes harm to the body. As a result, physiological and functional damage accumulates, setting off the onslaught of chronic inflammation and disease. Impaired energy metabolism alone can lead to changes in insulin sensitivity that affect a whole range of normal functions, from neural to sensory (especially visual and auditory) [6]. It must be stipulated that cellular senescence and aging are not transposable concepts. Aging is a process that is inherently time-dependent, whereas senescence occurs throughout life, even before birth during embryogenesis.
Recent advances in our understanding of the factors and mechanisms that cause and sustain cellular senescence may lead to the selective removal of senescent cells. The term senotherapy has been used to describe this unique approach [7]. Senotherapeutics have been proven to reduce the quantity of naturally existing senescent human cells in vitro, reverse or prevent senescence hallmarks, and improve physical and cognitive function, lengthening the lifetimes of aged mice [8].
2. Hallmarks of Senescence
The hallmarks of senescence are a set of characteristics or alterations acquired by cells as they transition to senescent states, e.g., morphological changes. A “biomarker” (an amalgamation of “biological marker”), in its broadest sense, is a quantifiable measure of a certain biological parameter or condition that can be measured with a certain degree of accuracy and reproducibility. Biomarkers are often associated with hallmarks, e.g., wide and flattened cells, which are framed within morphological changes. Stephen Naylor defined a biomarker as “an umbrella coalescence term” and, more precisely, “a characteristic that is objectively measured and evaluated as an indicator of normal biological or pathogenic processes” [50]. Regarding the interpretation of biomarkers of aging, one of the first definitions was proposed by Baker et al. in 1988: a “biomarker of aging is a biological parameter of an organism that either alone or in some multivariate composite will, in the absence of disease, better predict functional capability at some late age, than will chronological age” [51].
2.1. Stable Cell Cycle Arrest
Eukaryotic cell division is directed by a group of heteromeric serine/threonine kinases known as cyclin-dependent kinases (CDKs) [52]. The basic mode of action is the consecutive activation of each member of the family network, bringing about the phosphorylation of appropriate substrates and thus the passage through each step of the cell cycle (Figure 1).
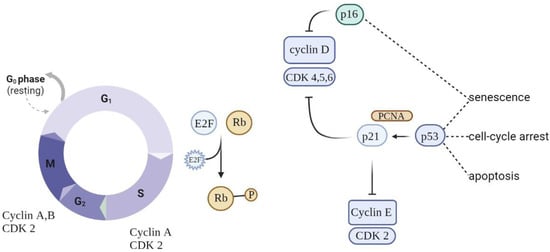
Figure 1. Schematic of cell cycle regulation (partial). To progress through the G1 phase, cyclin and CDKs should be set up and further phosphorylated. CDK inhibitors (CDKIs) halt cyclins from acting as cell cycle breaks. A close connection between cyclins, CDKs, and CDKIs is essential for correct cell cycle progression.
Cell cycle arrest is prompted by the p53/p21WAF1/CIP1 and p16INK4A/pRB tumor suppressor routes. p21WAF1/CIP1 acts downstream of p53, while p16INK4A acts upstream of pRB. Prolonged stress conditions may be a potential trigger for the activation of p16INK4a—a cell cycle regulator that acts as an inhibitor of CDK4/6 kinases [53]. This leads to arrest in the phosphorylation of RB, which in turn contributes to enduring cell cycle arrest. As long as p16INK4a is expressed, Rb proteins remain in a sustained hypophosphorylated state, stimulating attachment to E2F and cell cycle exit in G1 [54]. The overexpression of p16INK4a was detected in aging human skin, indicating a possible link between this suppressor protein and aging and senescence. Moreover, the upregulation of p16INK4a was observed in senescent fibroblasts in response to oxidative and DNA-damaging stressors.
When p53 is switched on, it acts as a regulator of the growth-suppressive transcriptional process by activating the cyclin-dependent kinase inhibitor gene p21, indirectly causing the hypophosphorylation of RB and cell cycle arrest [55,56]. Nevertheless, these genes are not absolute markers: the persistent activation of p21CIP1 (relevant in the launching of the senescence process) is not always observed in senescent cells, making it unreliable as a single marker [57]. At the same time, p53 is a regulator of apoptotic cell death, making it a dubious aid in discriminating between senescent cells and those undergoing apoptosis [58].
2.2. Metabolic Changes
In 1995, Dimri et al. detected the senescence-associated β-galactosidase (SA-β-gal) enzyme with optimum levels at pH 6 expressed by senescent cells; since then, it has become the best-known and most studied biomarker of cell senescence [59]. As cells age, their defense mechanisms become further corrupted, leading to amassed molecular debris [60], often associated with the altered function of lysosomal and proteosomal enzymes [61]. Detecting switches in normal metabolic function is a useful way to identify senescent cells. An increase in SA-β-gal is a biomarker that reflects the increase in the number and size of lysosomes [40]. SA-β-gal is not detected in quiescent or differentiated cells [40], yet it has been found in cells with intrinsically high lysosomal β-galactosidase activity, such as macrophages and postmitotic cells [62], and in several types of cancer cells [63]. Hence, the choice of SA-β-gal as an exclusive marker may lead to a false-positive outcome and requires the amplification of results using auxiliary markers.
A more recent enzymatic marker for senescence-associated lysosomal expansion, α-fucosidase, was found to be overexpressed in multiple senescent cell models [64]. Additionally, the attenuation of α-fucosidase was established to negatively affect the onset of senescence [65].
The enlargement of lysosomal substances in senescent cells is largely attributed to the accumulation of lipofuscin (LF) [66]. LF is represented by nondegradable yellowish-brown pigment granules that are mainly composed of an autofluorescent mixture of oxidized lipids, cross-linked proteins, oligosaccharides, and metals. This aggregate is a derivative of the oxidative and polymerization reactions between a wide range of cellular structures and macromolecules [67]. Because of its convoluted chemical structure, LF cannot be eliminated, causing it to accumulate in the lysosomes or cellular cytoplasm of lingering postmitotic and senescent cells that remain after normal autophagy. Over time, this leads to the extension of the lysosomal lumen to accommodate the ever-increasing amounts of LF. In contrast, proliferation-competent cells systematically attenuate the volume of LF during cell division [68]. For this reason, LF is commonly linked to aging and is referred to as the “age pigment” [69]. LF can be detected by microscopy techniques such as transmission electron [66] or confocal fluorescence microscopy [68].
Generally, metabolic changes in cell senescence are denoted by an increase in the ratio of AMP/ADP to ATP, leading to the activation of AMP-activated protein kinase (AMPK) signaling. The upregulation of AMPK causes a shift from a biosynthetically driven metabolism into a catabolism mode. Aging-related diseases are greatly impacted by the ability of AMPK activation to slow or stop the aging process. By the same token, in skin, senescence has been shown to affect various metabolic pathways in both dermal and epidermal cells, leading to deficiencies in key metabolites and protective proteins, which weakens the skin’s barrier function [70].
As a byproduct of mitochondrial metabolism, ROS are continuously created and removed by antioxidant mechanisms. To function properly, respond to metabolic stress, and avoid cellular senescence, ROS produced by mitochondria must be controlled. Mitochondrial ROS are a physiological activator of AMPK, and this activation results in an antioxidant response, reducing the amount of mitochondrial ROS produced. However, AMPK-deficient cells exhibit elevated amounts of mitochondrial ROS and develop premature senescence. These findings accentuate the critical role of AMPK in detecting and neutralizing mitochondrial ROS to maintain cellular metabolic equilibrium and resilience to stress and senescence [71].
2.3. Morphological Changes
During the transition to the senescent state, cells undergo a few changes in their morphological features [72]. The most obvious are the atypically distended dimensions and flat shape [73]. Cells exhibit substantial vacuolation and in some cases have multiple nuclei [74]. Moreover, adherent cells appear flattened, disarranged, and show random orientation in the culture plate [75].
Senescent cells often exhibit an augmented nucleus [76], which has been attributed to a range of factors, including a reduction in lamin B1. Lamin B1 is an intermediate filament protein incorporated into the inner portion of the nuclear envelope [77]. It aids in the stability of the nucleus, the replication of DNA, gene transcription, and cell proliferation. A lamin B1 deficit is part of the senescence-induced restructuring of the nuclear architecture. It is accompanied by the refashioning of chromatin organization, the loosening of heterochromatin, and the resulting enlargement of the nucleus [78,79].
Senescent cells exhibit a substantial increase in the number of mitochondria, and these organelles present an enlarged, distended shape. The enlargement of mitochondria can be attributed to the cell compensating for dysfunction by fusion and reduced division. Due to reduced autophagy in senescent cells, poorly functioning mitochondria are not cleared but rather accumulate in the cell. This amassing leads to a decrease in the mitochondrial membrane potential, accelerating the production of ROS. In addition, it has been observed that ROS have a damaging effect on mitochondrial DNA, which exacerbates organelle dysfunction. This is particularly evident in cases of photoaged skin, where mitochondrial DNA has a high level of mutations due to exposure to UVA compared to protected skin [80].
2.4. Epigenetic Alterations
Chromatin reorganization is a prominent genome-wide alteration in cells experiencing senescent transformation, which contributes to persistent proliferation arrest and transition into full senescence [81]. Senescent cells exhibit regions of condensed heterochromatin observed under microscopy [82]. These regions, known as senescence-associated heterochromatin foci (SAHF), are densely organized patches of DNA containing a constitutive heterochromatin marker histone H3 (H3K9me3—trimethylated at Lys9) in the core surrounded by the facultative heterochromatin marker H3K27me3 (trimethylated at Lys27).
Constitutive heterochromatin proteins, namely, di- or trimethylated forms of histone H3 (H3K9me2/3) and HP1 proteins associated with gene silencing, are easily detectable by immunofluorescent techniques, which highlight their role as potential senescence markers [83]. Notably, SAHFs do not carry sites of active transcription; thus, SAHFs are true transcriptionally inactive patches of heterochromatin. SAHFs are routinely identified by staining DNA with specific dyes such as DAPI, with senescent cells exhibiting spotty staining and “normal” nonsenescent DNA showing uniform staining [84].
Altered redox mechanisms have been observed to cause the general hypomethylation of the genome and the specific hypermethylation of DNA promoters, although it is not clear whether ROS-induced changes in the epigenetic makeup are exclusively a cause or a consequence of aging. Broad-based DNA hypomethylation has been shown to be strongly implicated in the expression process of aging genes, which is supported by the fact that cancer, a high-risk age-associated disease, shows the most pronounced effects of ROS-dependent DNA methylation. In aging, genomic regions with the activating histone post-translational modification H3K4me1 are more likely to have hypomethylated DNA sequences. The duality of the hyper- and hypomethylation processes of the different parts of the DNA reveals the complexity of the genomic mechanisms involved in cell senescence [85]. Nevertheless, aberrant DNA methylation could be both a potential biomarker and a tool to evaluate therapeutic treatments.
2.5. DNA Damage and Persistent DNA Damage Response
DNA damage, especially double-strand breaks (DSBs), is an integral aspect of senescent cells. DNA damage response in senescent cells is most often associated with proteins such as γH2AX (phosphorylated at Ser139) and an adjacent p53-binding protein 1 (53BP1), which are known to aggregate at DSBs. The breaking of the double strand activates the recruitment pathway of the ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR) protein kinases to the site of damage. These kinases are capable of converting histone H2AX to its phosphorylated form γH2AX, which quantitatively correlates with DSBs. ATM is known to phosphorylate a variety of substrates, particularly the checkpoint kinases CHK1/2, facilitating a downstream phosphorylation cascade and the activation of the p53/p21 signaling pathway [86]. The simultaneous detection of γH2AX and p53/p21 could be a viable option for detecting senescent cells.
Cellular senescence is also marked by the elevated expression of the promyelocytic leukemia protein (PML) [87]. PML is a normal constituent of the nucleus of most cell lines and acts as part of cell cycle regulation through the Rb and p53 pathways, which tend to accumulate in PML nuclear bodies. The extent to which PML accumulates in regions of unprocessed, unrepaired DNA correlates positively with the extent of DNA damage signaling, indicating that the transition to the senescent state may be associated with the corruption of the cell’s DNA repair system [88].
Another noteworthy epigenetic change connected to senescence is the formation of so-called DNA-SCARS (DNA segments with chromatin alterations reinforcing senescence). These foci appear to be a universal feature of most types of senescence, showing coupling with PML core bodies as well as an accumulation of activated forms of p53, ATR, and ATM [89,90]. Thus, these persistent foci could be considered excellent senescence comarkers.
Telomere dysfunction-induced foci (TIF) are an alternative version of DNA-SCARS, specifically located at uncapped telomere sites. These markers have been shown to concentrate in both senescent cells and aging tissues, as determined by the colocalization of 53BP1 and γH2AX at the telomeric ends of DNA [91]. Studies have associated a longer telomere length with decreased cellular senescence. Mice with hyper-long telomeres have been shown to express lower levels of global DNA damage, telomere-induced DNA damage, and p21, revealing the relationship between cellular senescence and telomeric length [92].
2.6. Apoptosis Resistance
Senescent cells employ a number of pathways to evade apoptosis [93]. Major survival pathways include ephrins and the Bcl-2 protein family, which act by actively suppressing apoptosis. Exemplary research conducted on murine models that studied the inhibition of antiapoptotic Bcl-2, Bcl-W, and Bcl-xL proteins showed apoptosis and the subsequent elimination of senescent cells [94]. Escape from apoptosis could also be achieved by the overexpression of p21, which appears to be a substantial inhibitor of p53-dependent apoptosis. Additionally, higher levels of p21 impede the activation of the c-Jun amino-terminal kinase (JNK) and caspase networks, both of which have been implicated in the apoptosis process. The dentification of these Bcl-2 proteins is deemed to be a convenient technique for localizing senescent cells. However, the upregulation of the synthesis of these markers is not limited to senescent cells; blood cells also show the overexpression of anti-apoptotic regulators [95]. While these proteins seem appealing as markers that are usually targets for senolytic agents, their expression is rarely chosen to assess this cell state [96]. Rather, evidence of the absence of annexin V and cleaved caspase-3 is regularly used as a marker to rule out apoptosis as a stress feedback process.
2.7. Secretory Phenotype
The SASP is one of the most thoroughly described hallmarks of the senescent state. Essentially, it is the ability of senescent cells to send inflammatory signals to neighboring cells in a paracrine manner (Figure 2). Among the several dozen identified factors that are secreted in a cell- and tissue-dependent manner, a number of marker molecules are commonly expressed by most senescent cells.
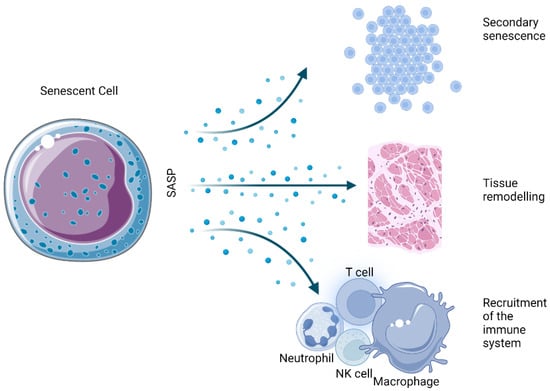
Figure 2. Physiological effects derived from the SASP of senescent cells.
The chemical composition of the SASP is highly dependent on the type and strength of the stimulus triggering senescence as well as the type of cells implicated in the process. For example, oncogene-induced senescence shows the exaggerated secretion of typical proteins in comparison to replicative or irradiation-induced senescence [97]. Moreover, when examining tissue and tumor material for SASP influences, it is very important to consider that the immune cell infiltrate and the degree of senescent cell accumulation could present additional unpredictable variables [98].
Even so, some crisscrossing has been demonstrated between a set of SASPs, with several proteins detected more or less universally. A basic SASP series consists of soluble molecules such as growth factors (IGFBPs, VEGFs, PDGFs, and HGFs) and interleukins [99]. The leading cytokine in the SASP process is proinflammatory IL-6, which appears to be directly driven by sustained DNA damage in keratinocytes, melanocytes, and epithelial cells, among others [100]. Another notable interleukin upregulated by senescent cells is IL-1, with both IL-1α and IL-1β overexpressed by various cell types [101].
In addition to the secretion of some proinflammatory factors, senescent cells also express enzymes for ECM remodeling, such as matrix metalloproteinases (MMPs), especially MMP1/3/9, which are involved in the breakdown of matrix proteins, serine/cysteine proteinase inhibitors (SERPINs), and tissue inhibitors of metalloproteinases (TIMPs) [45,102].
SASP-associated proteases greatly influence cell homeostasis by solubilizing membrane-associated proteins, breaking up and subsequently degenerating signaling molecules, processing, remodeling, or degrading the ECM [103]. These activities are responsible for the high potency of senescent cells in altering surrounding tissues.
SASPs have long been shown to be promising markers of cellular senescence. Furthermore, SASPs can be qualified and quantified both directly and indirectly by observing their known effects on surrounding cells. Nevertheless, it is important to understand that there are some major limitations inherent to secretory markers. SASPs can vary between cell types and different stages of senescence, and there is a gap in technology that does not allow for the high-throughput analysis of single-cell secretory phenotypes, which would be necessary to isolate a population on this basis [104].
2.8. Reactive Oxygen Species
ROS are well-known mediators of the senescence process. The generation of hydrogen peroxide, superoxide anions, and hydroxyl radicals disrupts normal cellular processes, induces senescence, and even leads to cell death [105]. Although the fact that ROS-induced DNA strand damage favors the onset and maintenance of senescence [106] has long been observed, the focus has currently shifted to the role of ROS as a signaling molecule in senescence induction. Signaling pathways closely involved in the senescence process, such as p53, p21, and p16, have been known to prompt ROS production [107], consequently promoting the upregulation of SASP factors [108]. Underscoring this role, McCarthy et al. 2013 showed that antioxidants and low oxygen tension restricted the production of IL-1α and downstream IL-6 and IL-8 [109].
Increasing inflammatory conditions accelerate the production of ROS in mitochondria through the recruitment of the cytokines TNF-α and IFN-γ [110]. Moreover, oxidative stress inhibits sirtuin activity, leading to higher levels of inflammation by inhibiting the superoxide dismutase enzyme (SOD) and preventing the inhibition of proinflammatory cytokines [111].
There is a clear relationship between cellular ROS levels and senescence. Low ROS levels are important for maintaining redox homeostasis and adequate antioxidant response. However, the increased accumulation of ROS triggers a variety of cellular responses, leading to irreversible cell cycle arrest that may turn into SASP, affecting surrounding tissue or apoptosis and subsequent cell death. Figure 3 shows the different cellular interactions related to senescence depending on the different intracellular levels of ROS.
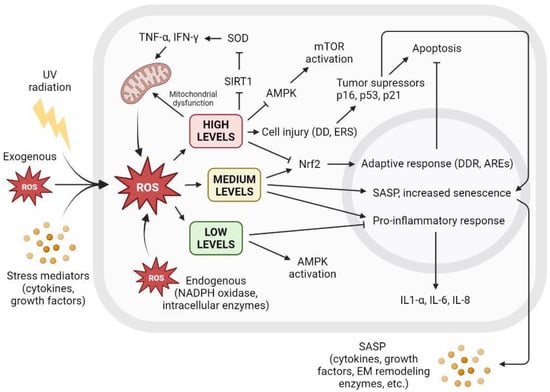
Figure 3. The level of intracellular ROS triggers adaptive response, apoptosis, or SASP. Intracellular ROS levels can vary depending on several factors, including extracellular factors such as exposure to UV radiation, high levels of oxidative stress, or proinflammatory environments with the presence of cytokines or growth factors. ROS are also produced intracellularly as part of normal cellular processes or at elevated levels derived from dysfunctional mitochondrial activity, as well as increased by NADPH oxidase activity or other intracellular enzymes. Low ROS levels activate AMPK and inhibit proinflammatory response. Low ROS levels do not promote cell senescence and contribute to redox homeostasis. Medium ROS levels activate Nrf2 and promote adaptive cellular response to fight ROS and prevent apoptosis, activate proinflammatory response, and increase SASP and its paracrine signaling, promoting cell senescence. High ROS levels cause mitochondrial dysfunctions that generate more ROS; inhibit AMPK, resulting in mTOR activation; and cause widespread cell damage by increasing cellular stress and the concentration of tumor suppressors and cell cycle arrest proteins, leading to apoptosis. AREs: AU-rich elements; AMPK: AMP-activated protein kinase; DD: DNA damage; DDR: DNA damage response; EM: extracellular matrix; ERS: endoplasmic reticulum stress; IFN-γ: interferon gamma; IL: interleukin; mTOR: mammalian target of rapamycin; NADPH: nicotinamide adenine dinucleotide phosphate; ROS: reactive oxygen species; SIRT1: sirtuin 1; SOD: superoxide dismutase; TNF-α: tumor necrosis factor alpha; UV: ultraviolet.
Inflammaging was introduced and extensively studied by C. Franceschi, who hypothesized that aging might be connected with an overall chronic increase in mediators of inflammation of a diverse nature [12]. This is presumably due to prolonged exposure to harmful substances during the lifespan or to fluctuations in the gut microbiota and other metabolic disturbances. Inflammaging is an ever-changing process that is easily transmitted by soluble factors to neighboring cells or even systemically [112]. Additionally, inflammaging leads to the chronic activation of the local immune system, resulting in persistent low-grade inflammation, creating a positive response loop with the immune system that affects its normal function. The aging process leads to noticeable changes in immune cells, such as the altered expression of surface markers, the weakening of the protective capacity, and a shift in the balance to the side of proinflammatory cytokine secretion. This acquired phenotype, referred to as “immunosenescence”, adds to the build-up of molecular damage in tissues; exacerbates various conditions; and (notably) significantly reduces the efficiency of the protective response against infection, tumors, and other damage [113].
In chronic cutaneous inflammation, the abnormal accumulation of molecular mediators has been associated with age-related changes in macrophage function [114]. The ability of these cells to carry out surveillance and clearance in an aging body becomes increasingly impaired, which is a key feature of immunosenescence. Subsequently, this “dereliction of duties” leads to an increase in overall oxidative stress and the further promotion of inflammation in the skin milieu.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12020444
This entry is offline, you can click here to edit this entry!