Mitochondria are key players in energy production, critical activity for the smooth functioning of energy-demanding organs such as the muscles, brain, and heart. Therefore, dysregulation or alterations in mitochondrial bioenergetics primarily perturb these organs. Within the cell, mitochondria are the major site of reactive oxygen species (ROS) production through the activity of different enzymes since it is one of the organelles with the major availability of oxygen. ROS can act as signaling molecules in a number of different pathways by modulating calcium (Ca2+) signaling. Interactions among ROS and calcium signaling can be considered bidirectional, with ROS regulating cellular Ca2+ signaling, whereas Ca2+ signaling is essential for ROS production. In particular, we will discuss how alterations in the crosstalk between ROS and Ca2+ can lead to mitochondrial bioenergetics dysfunctions and the consequent damage to tissues at high energy demand, such as the heart. Changes in Ca2+ can induce mitochondrial alterations associated with reduced ATP production and increased production of ROS. These changes in Ca2+ levels and ROS generation completely paralyze cardiac contractility. Thus, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells. These interplays in the cardiovascular system are the focus of this review.
- calcium
- ROS
- mitochondria
1. Introduction
2. ROS
The Alpha and Omega of Mitochondrial ROS
3. Calcium
3.1. Influx and Efflux of Ca2+ in Mitochondria
3.2. Key Ca2+ Targets and Roles in the Regulation of Mitochondrial Bioenergetics
4. The Interplay between Ca2+ and ROS
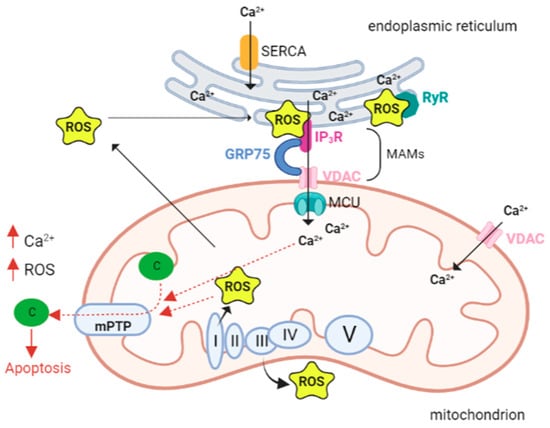
- Cardiac Muscle, ROS, and Ca2+ Signaling
The cardiomyocyte is a cell type that requires high amounts of energy to function efficiently. For these cells, ATP production by oxidative phosphorylation is essential, especially during contraction, after the increase in beating frequency/speed, and for all energy-intensive processes. About one-third of the volume of cardiomyocytes is occupied by mitochondria, which play a crucial role in the efficient coupling between energy production and cell requests [72].
During cardiomyocyte development, the sarcoplasmic reticulum (SR) and mitochondria undergo profound changes in coupling, shape, and distribution. In myoblasts (poorly differentiated muscle cells) mitochondria are few and elongated, much like a reticulum, while in myotubes (differentiated muscle fibers) they appear more spotted and globular. In addition, in myoblasts, mitochondria are coupled to Ca2+ release from the ER; in contrast, in myotubes, Ca2+ release is driven by SR. in this context, SR is crucial in the regulation of cytoplasmic calcium dynamics and cellular activity.
During differentiation, SR progressively change its shape as a beehive with tubular structure [84]. Ca2+ transfer between SR and mitochondria is particularly important in cardiac muscle, where ATP demand is high due to the energy required for excitation–contraction coupling (ECC). Ca2+ is uploaded into the mitochondria to invigorate metabolism, generate the ATP needed for contraction, and mediate Ca2+ elimination from the cytosol in the relaxation phase [85].
During ECC, the increase in intracytoplasmic calcium required for contraction induction is generated by Ca2+-dependent activation of ryanodine receptors (RyRs). These are Ca2+ release channels located on the SR, which is the main intracellular storage of this ion. In cardiomyocytes both RyRs and inositol 1,4,5-trisphosphate receptors (IP3Rs) are present. In myoblast mitochondria, Ca2+ release from the ER is mediated by IP3R. During cell differentiation, the expression of RyRs (particularly isoform 2) increases dramatically in cardiac muscle, and this is critical for excitation–contraction coupling. Simultaneously, IP3Rs play different roles, such as regulating gene transcription and hypertrophy [84,86], although the physiological contribution of IP3R-mediated Ca2+ release is not completely elucidated [85].
As it is the case in other cell types, also in cardiomyocytes the transfer of Ca2+ from SR to mitochondria involving IP3Rs in MAMs is mediated by the VDAC1-GRP75 (a mammalian heat shock stress protein 70). In this context, IP3R1 is called up, binds VDAC1 through GRP75 that tethers the two proteins by binding to their regions exposed in the cytosol and therefore forming a channel complex through which there is Ca2+ transfer between SR and mitochondria, [87].
5.1. Calcium and ROS in Heart Failure
Mitochondria and SR are interrelated and connected, and Ca2+ is crucial for the functioning of both organelles. Mitochondrial dysfunction is associated with alterations in Ca2+ levels [87–95].
Dysregulated calcium handling is a hallmark in cardiac dysfunction. In particular, changes in Ca2+ can induce mitochondrial alterations with reduced ATP production and increased production of ROS. Thus, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells [96–101]. Different studies reported a correlation between an increased level of ROS (in plasma and heart) and the gravity of left ventricular dysfunction [102–104].
Heart failure induces the activation of a number of mechanisms that result in a reduction in mitochondrial calcium transfer. Specifically, there is an increase Ca2+ influx in through the NCLX due to increased Na+ in the cytosol, reduction in MCU opening, SR Ca2+-ATPase activity, and ryanodine receptor expression. The final combined effect of these events is the negative alteration of the Krebs cycle [105–110]. As a result, the accumulation of oxidized pyridine nucleotides prevents the production of ATP from NADH and the detoxification of ROS by NADPH. The antioxidative enzyme responsible for the regeneration of NADPH from NADH is the nicotinamide nucleotide transhydrogenase (NNT) [111]: under pathological conditions, when the metabolic demand increases, the direction of the NNT reaction is reversed, oxidizing NADPH to regenerate NADH and produce ATP, and interfering with the NADPH-dependent antioxidative capacity [10]. Because mitochondria represent the main ROS scavenging system of the cell [103], the oxidation of NADPH by NNT could cause excessive mitochondrial ROS release, that leads to necrosis, left ventricular dysfunction, and death [112].
5.2. Mitochondrial ROS vs. ER ROS
Mitochondria have been considered the primary source of ROS [7,113–118], but several lines of evidence propose an important role also for ROS generated in the ER in cardiovascular diseases.
MAMs are the contact sites between the endoplasmic reticulum or sarcoplasmic reticulum. Our knowledge of calcium signaling in cardiac pathologies, where ER and oxidative stresses are predominant [5,119–121], suggests that calcium may, in fact, be the cause, rather than the effect, of altered mitochondrial ROS. This implies that calcium overload signals mitochondria to produce lethal levels of ROS.
The ER is a primary site of protein synthesis and post-translational processing, and the protein folding process is influenced by the redox state of the ER. A well-known condition of the ER, called ER stress, due to the accumulation of misfolded polypeptides, is caused by the accumulation of oxidant equivalents in the ER. ER stress activates the unfolded protein response (UPR), which raises the folding protein capacity, resulting in an increased production of oxidative equivalents and an additional deterioration of the redox state [122]. During ER stress calcium channels, both ryanodine and inositol-3-phosphate receptors open [123] with the release of calcium, which is crucial for the contraction of the muscles. To sustain Ca2+ homeostasis, Ca2+ returns to the SR/ER through SERCA (sarco-endoplasmic reticulum calcium ATPase). In pathological conditions, Ca2+ homeostasis in the ER is dysregulated, with an enhanced Ca2+ release from mitochondria [124,125]. Ca2+ within the mitochondria generates superoxide, a marker for oxidative stress [126]. Cardiac contractility is entirely paralyzed by Ca2+ leakage, Ca2+ overload, and ROS generation.
5.3. In Vivo Mouse Model of Postmyocardial Infarction
Type 2 ryanodine receptor (RyR2) and type 2 inositol 1,4,5-trisphosphate receptor (IP3R2) [127–129] are the two intracellular Ca2+ channels in SR of cardiac cells, and the RyR2 receptor is crucial for cardiac excitation–contraction (E–C) coupling in cardiomyocytes.
In several murine models of postmyocardial infarction that have been generated so far, it has been shown that, based on the physical and functional association between the SR and mitochondria, when the SR calcium was released in cardiomyocytes by the two main channels, it accumulated in mitochondria, bringing to mitochondrial dysfunction, oxidative stress and decreased ATP production [88,130,131].
To investigate whether Ca2+ accumulation in mitochondria of failing hearts was caused by Ca2+ leak through the RyR2 receptor, Santulli et al. generated a murine model carrying a homozygous RyR2 mutation that renders the channel leaky (RyR2S2808D/S2808D) and a mouse model with a homozygous RyR2 mutation that renders the channel protected against leak (RyR2S2808A/S2808A) [129]. In cardiomyocytes derived from the RyR2S2808D/S2808D mice, there was an increased mitochondrial Ca2+ and ROS production, the presence of dysmorphic and malfunctioning mitochondria, a reduction in mitochondrial size, and low fusion-to-fission ratio compared with wild-type (WT) and RyR2S2808A/S2808A cardiomyocytes [129].
In opposition, cardiac-specific deletion of the IP3R2 receptor did not produce consequences on mitochondrial Ca2+ accumulation, as observed in the murine model (IP3R2CVKO), in which IP3R2 expression was suppressed in ventricular cardiomyocytes. IP3R2CVKO mice survived to adulthood without showing alteration in mitochondrial function. Ca2+ sparks, SR Ca2+ load, mitochondrial Ca2+ level, and ROS production were not affected in IP3R2CVKO ventricular cardiomyocytes. Moreover, IP3R2CVKO mice exhibited normal myocardial mitochondria and no significant effect after postmyocardial infarction [129].
5.4. Drug Targeting of Mitochondrial Ca2+ and Homeostasis
Based on the observed experimental data, the mitochondrial redox state might be a plausible drug target in cardiac alterations. Two distinct approaches have been used to modulate ROS and Ca2+ in mitochondria: (i) direct targeting of mitochondrial ROS by agents that accumulate in mitochondria; (ii) normalization of mitochondrial Ca2+ signaling to guarantee a balance between mitochondrial ROS emission and detoxification.
For the direct targeting of mitochondrial ROS, drugs that have been developed to accumulate in mitochondria or scavenge ROS (MitoQ) or directly act at the ETC level to reduce ROS production (SS-31). As an example, CGP-37157 is a benzodiazepine compound selectively inhibiting NCLX, the mitochondrial Na+/ Ca2+ exchanger by which mitochondrial Ca2+ is extruded. CGP-37157 suppresses mitochondrial Ca2+ efflux, abolishes NADH oxidation, and reduces ROS production in failing cardiomyocytes [110]. Overall, experimental studies performed with CGP-37157 showed the positive effects of mitochondrial NCLX inhibition in animal models of heart failure and in isolated heart cells. However, currently, there are no clinical trials to test the effect of these drugs.
A different strategy to normalize mitochondrial Ca2+ signaling is to reduce the concentration of intracellular Na+. In heart failure, [Na+]i is increased [132], and during excitation–contraction coupling, it hinders mitochondrial Ca2+ accumulation since it increases mitochondrial Ca2+ efflux via the NCLX [102]. It has been shown that an elevated late Na+ current (late INa) is required to increase cytosolic Na+. An enhanced late INa has been related to elevated mitochondrial ROS emission. This, in turn, worsens late INa through Ca2+-/calmodulin-dependent kinase II (CaMKII) oxidation. CaMKII [133,134] can enhance late Na+ current and cause arrhythmias during HF [135].
Ranolazine is a piperazine derivative inhibitor of late Na+ current [136] that was approved by the FDA in 2006 for chronic angina and cardiac ventricular dysrhythmias [137]. MERLIN-TIMI 36 trials provided the hypothesis that ranolazine may be particularly beneficial in patients with HF [138]. In this trial, patients with acute coronary syndrome profited from ranolazine, particularly when they had elevated levels of brain natriuretic peptide as a marker of HF [138]. Evidence suggested that the mechanisms of action could be the blocking of the late Na+ current that occurs during ischemia, the blocking of mitochondrial complex I activity, or by modulating mitochondrial metabolism [139]. In mitochondria isolated from the hearts of patients treated with ranolazine, there was decreased cytochrome c release and mild resistance to the opening of mPTP when compared with control hearts. Through its late Na+ current blocking action, ranolazine protected the heart during IR injury by decreasing the load of cytosolic (c)Ca2+ and mitochondrial (m)Ca2+. The final effect was a reduction in necrosis and apoptosis [139].
Recently, empagliflozin has demonstrated its positive effects in regulating cytosolic Na+ and mitochondrial Ca2+. It selectively inhibits the sodium glucose cotransporter 2 channel (SGLT-2) [140], which reabsorbs the filtered glucose in the renal early proximal tubule. Its main role is to reabsorb the majority of filtered glucose, decreasing renal glucose reabsorption and, thus, hyperglycemia. A clinical trial in 7020 patients, the EMPA-REG OUTCOME study, showed that patients with type 2 diabetes at high cardiovascular risk and treated with empagliflozin showed ameliorated cardiovascular endpoints and reduced death from HF when the study drug was added to standard care [141,142]. Empagliflozin might reduce [Na+]i in cardiac myocytes independently of SGLT inhibition, possibly by interacting with the Na+/H+ exchanger [143]. Thereby, this drug empowered mitochondrial Ca2+ in cardiac myocytes.
Antioxidants such as vitamins E and C have been used in animal experiments, but promising results were observed in preclinical studies only [144]. In subsequent clinical trials, none of these compounds led to any significant benefit. Despite evidence that mitochondria are crucial for oxidative damage, compartmentalization and the interaction between compartments probably explain why “generic” antioxidants, such as vitamins, failed to target dysfunctional mitochondria and altered Ca2+ levels.
A more targeted and specific approach has been focused on NOXs, which have the biological role of producing ROS for signaling purposes and transferring electrons across biological membranes to produce O2- [145]. Along with the already well-known lipid-lowering effect of statins in preventing cardiovascular disease, their antioxidant effect through NOX2 inhibition has been demonstrated. Statin treatment provided positive antioxidant effects in vitro in isolated cardiomyocytes and in animal models of HF [141,142]. However, in the CORONA study, comprising 5011 HF patients, statin therapy did not show to reduce cardiovascular death [146].
In January 2021, the FDA approved vericiguat, a new soluble oral guanylate cyclase stimulator, which enhances the production of cyclic guanosine monophosphate, for the treatment of chronic HF. It is well known that the cardioprotective pathway of soluble nitric oxide guanylate cyclase-cyclic guanosine monophosphate is compromised in patients with HF [147].
Based on data from the SOCRATES-REDUCED and VICTORIA trials in adult patients with chronic HF, this drug was approved for medical treatment. In the VICTORIA trial, the dose of vericiguat was initially 2.5 mg/day and increased after 2 weeks to 5 mg/day and then to 10 mg/day. Placebo doses were administered in the same manner. After about 1 year, 90% of patients were treated with the 10 mg target dose. The median follow-up for the primary endpoint was 11 months. The annualized absolute risk reduction with the drug treatment was ~4.2% over the course of the study [148]. The recommended initial oral dose of vericiguat was 2.5 mg/day, with food intake, and should be doubled approximately every 2 weeks to reach the target maintenance dose of 10 mg/day. Based on the VICTORIA trial, vericiguat significantly reduced the rate of hospitalization and cardiovascular death attributed to HF. In fact, regardless of patients’ atrial fibrillation status, vericiguat was better in preventing all causes of death, cardiovascular death, hospitalization, and HF.
The main advantage of this drug is that it avoids the risk of electrolyte imbalance or renal damage [149]. Moreover, vericiguat bypasses the many problems of current therapies, such as the gradual decline in effectiveness, drug dose-dependent tolerance, and off-target effects due to a lack of specificity, thus representing a game changer in the treatment of HF, because it has great potential to reduce the severity of the disease [150].
5.5. Cardiomyopathy with Mitochondrial Dysfunction-Associated Genes
Several signaling pathways, ion homeostasis, and metabolism are impaired in cardiomyopathies. Therefore, mitochondria, which are the cellular powerhouse, are involved in many of these processes. The known genes that are altered in cardiomyopathies with mitochondrial dysfunctions are reported in Table 1.
Table 1. Susceptibility loci and candidate genes linked to cardiomyopathies with mitochondrial alterations.
|
Chromosome Location |
Phenotype |
Phenotype MIM Number |
Inheritance |
Gene/Locus |
Gene/Locus MIM Number |
References |
|
|
6q13 |
Combined oxidative phosphorylation deficiency 10 |
614702 |
AR |
MTO1 |
614667 |
[151] |
|
|
7q34 |
Sengers syndrome |
212350 |
AR |
AGK |
610345 |
[152] |
|
|
4q35.1 |
Mitochondrial DNA depletion syndrome 12B (cardiomyopathic type) |
615418 |
AR |
SLC25A4 |
103220 |
[153] |
|
|
Mt |
Hypertrophic cardiomyopathy with kidney anomalies due to mtDNA mutations |
|
Mitochondrial inheritance |
MT-TL1 |
|
[154] |
|
|
mt |
Mitochondrial DNA- related cardiomyopathy and hearing loss |
|
Mitochondrial inheritance |
MT-TK |
|
[155] |
|
|
Xq28 |
Barth syndrome |
302060 |
XLR |
TAFAZZIN |
300394 |
[156] |
|
5.5.1. MTO1
MTO1 encodes for a mitochondrial protein involved in tRNA modification and protein synthesis and catalyzes the 5-carboxymethyl aminomethylation of the uridine base in mitochondrial tRNAs that transport Gln, Glu, and Lys. Mutations in this gene cause an autosomal recessive disorder known as combined oxidative phosphorylation deficiency 10 (COXPD10), characterized by altered OXPHOS activity. Alterations in mitochondrial oxidative respiration cause hypertrophic cardiomyopathy and lactic acidosis in early infancy and complications that can be fatal in severe cases [151].
5.5.2. AGK
The human AGK gene encodes for the mitochondrial acylglycerol kinase, an enzyme located on the mitochondrial membrane involved in the formation of phosphatidic and lysophosphatidic acids. Homozygous or compound heterozygous mutations in the AGK gene have been associated with Sengers syndrome, also known as cardiomyopathic mitochondrial DNA (mtDNA) depletion syndrome-10 (MTDPS10). This syndrome is characterized by skeletal myopathy, hypertrophic cardiomyopathy, exercise intolerance, lactic acidosis, and congenital cataracts. Skeletal muscle biopsies showed severe mtDNA depletion [152], and cardiomyopathy is the major cause of early death [157].
5.5.3. SLC25A4
SLC25A4 gene encodes for the Solute Carrier Family 25 Member 4, expressed in mitochondria. The function of this protein is to translocate ADP from the cytoplasm into the mitochondrial matrix and ATP in the opposite direction. Homozygous or compound heterozygous mutations in this gene cause mitochondrial DNA depletion syndrome 12B (MTDPS12B), an autosomal recessive mitochondrial disease. Onset occurs in infancy with progressive hypertrophic cardio- and skeletal myopathy. The presence of red and irregular fibers, accumulation of abnormal mitochondria, and mtDNA depletion can be observed in skeletal muscle biopsies from the affected individuals [153].
5.5.4. MT-TL1
MT-TL1 is a mitochondrial gene that encodes the t-RNA-Leu. Mutations in MT-TL1 can result in multiple mitochondrial deficiencies and associated disorders. Among them, variants in MT-TL1 cause hypertrophic cardiomyopathy with renal abnormalities due to altered oxidative phosphorylation. This syndrome is characterized by hypertrophic and dilated cardiomyopathy, myopathy with hypotonia with developmental delay, and/or regression with cerebral atrophy and chronic renal [154].
5.5.5. MT-TK
Another mitochondrial RNA gene is MT-TK, affiliated with the tRNA class, which transfers the amino acid lysine during translation. Defects in this gene are associated with mtDNA-related cardiomyopathy and hearing loss. The clinical manifestation of this rare mitochondrial disease is characterized by progressive sensorineural hearing loss along with hypertrophic cardiomyopathy and encephalomyopathy. Other symptoms might be present such as progressive external ophthalmoparesis (PEO), ataxia, slowed speech, myalgia, and muscle weakness [155].
5.5.6. TAFAZZIN
The TAFAZZIN gene encodes a protein expressed at high levels in cardiac and skeletal muscle. Barth syndrome is caused by variants in the TAFAZZIN gene [87–95,154]. This syndrome is an X-linked disease with dilated cardiomyopathy (CMD) with endocardial fibroelastosis (EFE), a proximal skeletal myopathy, growth retardation, neutropenia, and organic aciduria [156]. Hypertrophic cardiomyopathy, ventricular arrhythmia, motor delay, and other cardiac and motor symptoms might be present.
- Conclusions and Future Perspectives
In cardiac cells, ATP production by oxidative phosphorylation is essential, especially during contraction, after the increase in beating frequency/speed, and for all energy-intensive processes. As a consequence, dysregulation in mitochondrial bioenergetics severely affects their function. Changes in Ca2+ can induce mitochondrial alterations associated with reduced ATP production and increased production of ROS. In this context, ROS can hinder the excitation–contraction coupling, inducing arrhythmias, hypertrophy, apoptosis, or necrosis of cardiac cells [97–101]. The search for new methods to recover these damages has seen a great expansion in recent years through the development of functional biomaterials and biomimetic scaffolds in regenerative medicine, which finds fertile ground for the application of cardiac therapies, considering the low regenerative capacity of this tissue [158].
Many efforts have been made in order to develop functional cardiac tissue in vitro, especially with the development of knowledge in the field of pluripotent stem cells (PSCs). Human pluripotent stem cells (hPSCs) have the capacity to differentiate into different cell types. They can be derived from the cells within the blastocyst at the beginning of embryonic development, from which the various structures of the fetus originate [159], or they can be derived from human cells that are de-differentiated by expression of Yamanaka factors (Oct3/4, Sox2, Klf4, c-Myc), known as induced pluripotent stem cells (iPSCs) [160].
The use of iPSCs is an emerging application for the generation of disease models and the search for new therapies, making it possible not to use animal models and to overcome the ethical problems associated with human embryonic cells. The reprogramming of cells takes place in the context of the individual’s genetic background, and it has been shown how the genetic and epigenetic behaviors of iPSCs reflect those of the donor individual’s cells. Therefore, iPSCs have the potential to generate tissue-like cells or structures, such as cardiomyocytes, that are exactly those of the patient with the disease of interest, including the main inherited cardiomyopathies that we previously cited.
Branco et al. recently developed a protocol for the generation of self-organized human multilineage organoids that recreate the co-presence of several specialized cells [161]. Co-culturing these organoids together with cardiomyocytes, a functional cardiac organoid surrounded by myocardial-like tissue was generated, recapitulating the structure and function of mature cardiac cells [161].
Tenreiro et al. reported data from trials aimed at generating cardiomyocytes from cells derived from cardiomyopathy patients and studying their molecular mechanisms [162]. An important example of this approach is DMDstem (NCT03696628), a trial completed in 2021 and performed in children with genetic cardiomyopathy vs. healthy ones. However, the outcomes of this study are not yet reported in the literature. Another trial, IndivuHeart (NCT02417311), initiated in 2014 and completed in 2019, aims to ensure individualized early risk assessment for heart disease and to make engineered heart tissue technology (hiPSC-EHT) a clinically applicable strategy [163].
Advances in hiPSCs biology, genome-editing technologies, and cardiac tissue engineering all concur to generate healthy cardiac tissue from the patient himself, opening doors for targeted therapy and personalized medicine.
Author Contributions: Conceptualization, E.B. and C.D.; C.D., E.C.-S., and B.D.N. writing—original draft preparation, C.D. and E.B.; writing—review and editing, E.B.; funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding: This research was funded by the University of Bologna RFO2022 grant to E.B. C.D. is a recipient of the Fondazione U. Veronesi fellowship 2022. B.D. is a recipient of the Ph.D. program FSE REACT-EU -A.Y. 2021/2022.
Institutional Review Board Statement: Not applicable
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.
This entry is adapted from the peer-reviewed paper 10.3390/antiox12020353