Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Clinical Neurology
Glioblastoma (GBM) is the most aggressive and malignant primary brain tumor in adults. Despite multimodality treatment involving surgical resection, radiation therapy, chemotherapy, and tumor-treating fields, the median overall survival (OS) after diagnosis is approximately 2 years and the 5-year OS is poor. Considering the poor prognosis, novel treatment strategies are needed, such as immunotherapies. Natural killer (NK) cell-based immunotherapy involves the new feature of recognizing GBM via differing mechanisms from that of T cell-based immunotherapy.
- glioblastoma
- NK cell
- immunotherapy
1. Introduction
Glioblastoma (GBM) is the most common and aggressive primary brain tumor. The annual incidence of GBM is 3.19 per 100,000 [1] and it is classified as grade IV by the World Health Organization [2]. For several decades, the standard GBM therapy has consisted of maximum safety resection, adjuvant radiotherapy, and chemotherapy with temozolomide, termed the Stupp regimen. Despite the multidisciplinary therapy, the median overall survival (mOS) is only 15–17 months and the 5-year overall relative survival is only 5.8% [3,4]. Several novel strategies were investigated, where the addition of tumor-treating fields to standard treatment statistically significantly improved progression-free survival and OS [5]. A recent phase 2 trial of intratumoral oncolytic herpes virus G47∆ for residual or recurrent GBM demonstrated survival benefits and a good safety profile, which led to the approval of G47∆, albeit with conditions under the early approval system of Japanese-specific law, as the first oncolytic virus product in Japan [6]. Figure 1 summarizes the multimodality treatment against GBM. However, given the poor prognosis of patients with GBM, further novel approaches are needed for GBM treatment.
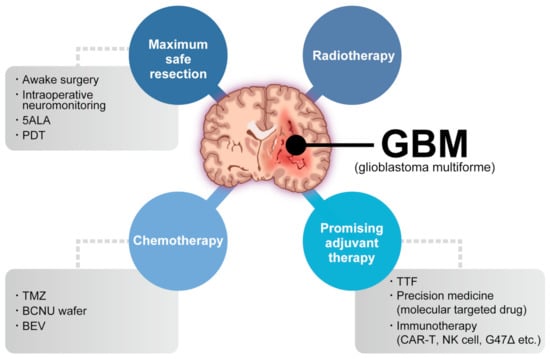
Figure 1. Multimodality treatment against GBM. While the standard therapy consists of maximum safety resection, adjuvant radiotherapy, and chemotherapy with temozolomide, which is collectively termed the Stupp regimen, several promising adjuvant therapies have been used for treating GBM. 5ALA: aminolevulinic acid, PDT: photodynamic therapy, TTF: tumor-treating fields, TMZ: temozolomide, BCNU: carmustine, BEV: bevacizumab.
Almost every immunotherapy mainly aims to generate a tumor-specific immune response to selectively eliminate tumor cells as a result of T cell activation. The representative immunotherapies are checkpoint inhibitor and chimeric antigen receptor (CAR) T-cell therapies, which have been used in other solid tumors and hematologic malignancies [7,8,9]. Contrastingly, several GBM immunotherapies have long been investigated, but few attractive strategies have been reported.
Compared to T cell-based therapy, natural killer (NK) cell-based therapy approaches tumors from new aspects. First, NK cells recognize the tumor, which consists of heterogeneous cells, via multiple activating and inhibitory receptors despite the diminished or absent expression of major histocompatibility complex class I (MHC-I) molecules [10]. Second, NK cells are important for recruiting conventional type 1 dendritic cells (cDC1s) and subsequently CD8+ T cells [11,12]. These functions promote cancer immunity cycle activation, which has the advantage of overcoming the immunosuppressive GBM tumor microenvironment (TME) [13,14].
2. NK Cell and NK Cell-Based Immunotherapy for Cancer
More than 40 years ago, it was determined that NK cells recognize cancer cells in mice and humans without antigen sensitization [58,59,60,61,62]. Recent research focused on the potential of NK cells in cell-based therapies. NK cells are considered an important part of the immune system, where they control microbial infections and tumor progression [58,63]. In patients and animal models, NK cell deficiency or impairment led to recurring virus infections and increased incidence of various types of cancer. In particular, NK cells controlled transplantable tumor growth and metastasis in numerous mouse models via antibody depletion of NK cells [64]. Additionally, NK cells are the founding members of the innate lymphoid cell (ILC) family [65]. In human peripheral blood, bone marrow, and tissues, NK cells can be identified by the expression of neural cell adhesion molecule (NCAM: CD56) and the absence of T cell receptor (TCR) and CD3 [66]. In the bone marrow, human NK cells derive from CD34+ hematopoietic progenitors and mature in the lymphoid organs [67,68]. Despite the differentiation from progenitor cells, NK cells persist in peripheral blood [69,70]. Human NK cell turnover in blood occurs over approximately 2 weeks [71].
NK cells can recognize tumor cells based on a balance between stimulatory and inhibitory receptors [stimulatory receptors: DNAX accessory molecule 1 (DNAM1), 2B4 (also known as CD244) and NK group 2D (NKG2D); inhibitory receptors: killer cell immunoglobulin-like receptors (KIRs), TIGIT, killer cell lectin-like receptor subfamily G member 1 (KLRG1), T-cell immunoglobulin mucin family member 3 (TIM3), and programmed death 1 (PD1)] [72,73] (Figure 2). In detail, the main activating receptors are natural cytotoxicity receptors (NCRs: NKp-46/NCR1, NKp44/NCR2, NKp30/NCR3) [74,75,76,77]. B7-H6 and BAG6/BAT3 represent NKp30 ligands [78,79]. NKp44 recognizes a specific human leukocyte antigen (HLA)-DP molecule (HLA-DP401) and PCNA [80,81]. Barrow et al. also reported that platelet-derived growth factor (PDGF)-DD engagement of NKp44 triggered NK cell secretion of interferon (IFN)-γ and tumor necrosis factor alpha (TNF-α), and a distinctive transcriptional signature of PDGF-DD-induced cytokines and the downregulation of tumor cell-cycle genes correlated with NCR2 expression and greater survival in glioblastoma [82]. Gaggero et al. identified the extracellular matrix protein nidogen-1 (NID1) as a ligand of NKp44 [83]. NKp46 binds to the soluble plasma glycoprotein complement factor P/properdin [84]. Garg et al. reported vimentin (a 57-kDA molecule) as a putative NKp46 ligand [85]. The lysis of influenza virus (IV)-infected cells is mediated by the interaction between NKp46, and the IV hemagglutinin (HA) type 1 expressed by the infected cells [86,87,88]. NKG2D is another important NK receptor that transduces activating signals from the transmembrane adaptor protein DAP10 and recognizes UL16-binding proteins (ULBPs) and MHC class-1 related chain (MIC) A/B [89].
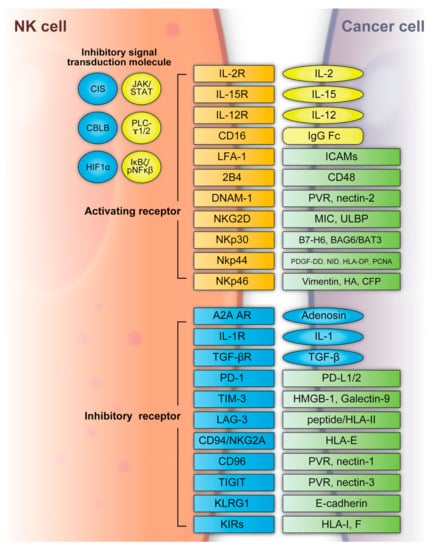
Figure 2. NK cell activating and inhibitory receptors. NK cells recognize tumor cells based on a balance between the stimulatory and inhibitory receptors above. A2A AR: A2A adenosine receptor, BAG6/BAT3: BCL2-associated athanogene cochaperone 6, CBLB: Casitas B-lineage lymphoma pro-oncogene-b, CIS: cytokine-inducible SH2-containing protein, DNAM1: DNAX accessory molecule 1, HA: IV hemagglutinin, HIF1α: hypoxia-inducible factor 1α, HLA: human leukocyte antigen, HLA-DP: human leukocyte antigen DP molecule, HMGB1: high-mobility group protein 1, ICAMs: intracellular adhesion molecules, IgG Fc: constant region of immunoglobulin, IL: interleukin, KIR: killer cell immunoglobulin-like receptors, JAK/STAT: Janus kinase/signal transducer and activator of transcription, KLRG: killer cell lectin-like receptor subfamily G member 1, LAG3: lymphocyte-activation gene 3, LFA-1: lymphocyte function-associated antigen-1, MIC: major histocompatibility complex class 1-related chain, NF-κB: nuclear factor kappa B, NID: nidogen, NKG2D: NK group 2D, NKp30: natural killer cell p30-related protein, NKp44: natural killer cell p44-related protein, NKp46: natural killer cell p46-related protein, PCNA: proliferating cell nuclear antigen, PD-1: programmed cell death 1, PDGF-DD: platelet-derived growth factor, PD-L1/2: programmed cell death ligand 1/2, PLC-γ1/2: phospholipase C γ1/2, PVR: poliovirus receptor, TGFβ: transforming growth factor β, TIGIT: T-cell immunoreceptor with immunoglobulin and ITIM domains, TIM3: T-cell immunoglobulin mucin family member 3, ULBP: UL16-binding protein.
Regarding inhibitory receptors, NKG2A–CD94 inhibits NK cell function when bound by HLA-E [90]. However, NKG2C–CD94 heterodimers activate NK cells when bound to HLA-E [91]. In non-HLA-specific inhibitory NK receptors, PD-1, TIGIT, CD96, TIM3, and CD161 function as NK cell activation immune checkpoints, and their ligands are PDL1, Poliovirus receptor (PVR)/PVRL2, galectin-9/high-mobility group protein 1 (HMGB1)/phosphatidylserine (PtdSer), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), and lectin-like transcript-1 (LLT1), respectively [92]. KLRG1 is another inhibitory receptor expressed by activated NK cells [93]. KLRG1 binds E-cadherin and inhibits human ILC2 function [92]. NK cells express several co-receptors that enhance NK cell triggering activity via NCRs or NKG2D, where the representative co-receptors are 2B4, DNAM-1, and NKp80 [94,95]. Using these receptors, NK cells can recognize whether the adjacent cell (infected or tumor cells) is targeted for killing without prior sensitization. NK cells eliminate cells with diminished or absent MHC-I expression [96]. The MHC-I ligand is a set of KIR inhibitory receptors, which suppress NK cell function and minimize the destruction of healthy self-cells [72,97]. NK cells undergo so-called licensing or education during their development to avoid self-reactivity [98]. NK cells chronically stimulated by self-ligands might become anergic if the inhibitory receptors do not mitigate the stimulation [99,100]. Although the ligation of self-MHC suppresses mature NK cells, the suppression is relieved if MHC is altered or downregulated, which may occur in tumor cells [99,100]. Additionally, the representative classical HLA-F inhibits NK cell function through KIRs [101]. NK cells also have a potent activator, CD16, which recognizes the constant region (Fc) of IgG antibodies and is responsible for antibody-dependent cell-mediated cytotoxicity (ADCC) [102,103].
NK cell functions are regulated by intracellular checkpoint molecules, which are inhibitory signal transduction molecules. Cytokine-inducible SH2-containing protein (CIS) is encoded by cytokine inducible SH2-containing protein (CISH) as an IL-15-inducible inhibitor of IL-15 signaling in mouse NK cells [104]. CIS acts as an intracellular checkpoint receptor in tumors with increased IL-15 concentrations [105]. Barsoum et al. reported that reduced nitric oxide levels in prostate cancer cells induced another intracellular checkpoint molecule, hypoxia-inducible factor 1α (HIF1α), which augmented a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) expression and significantly increased MICA secretion in the extracellular milieu [106]. Moreover, the expression of HIF-1α, a transcriptional factor, promotes multiple signaling and induces immune suppression, including that of NK cells [107,108,109,110].
The hypoxic TME contributes to immune escape in cancer. Casitas B-lineage lymphoma pro-oncogene-b (CBLB) interacts with its specific targets via phosphotyrosine-containing sequence motifs generated on activated protein tyrosine kinases that mediate activating signal transduction [111,112]. In NK cells, CBLB is activated and stabilized through inhibitory receptor signaling and reduces NK cell degranulation and cytotoxicity by targeted degradation of the adaptor protein linker for activation of T cells (LAT) [113,114].
Although NK cells are classified as innate cells, their responses can exhibit the adaptive phenotype of immunological memory or trained immunity under circumstances such as viral infections or stimulation with IL-12, IL-15, and IL-18 cytokines [115,116]. Recently, single-cell RNA sequencing analysis tracked pathogen-specific adaptation within the innate immune system via NK cell memory following human cytomegalovirus infection. NK cell clonal expansion and persistence within the human innate immune system were demonstrated in detail, where these mechanisms evolved independently of antigen-receptor diversification [117].
When NK cells encounter a target cell and are activated, a synapse is formed with the target cell and microtubules transport lytic granules, which converge towards the synapse [118]. Additional signals from the synapse lead to lytic granule polarization; the granules contain the key effectors of cytotoxicity: perforin and granzymes [119,120]. The release of a single granule is sufficient to kill a target tumor cell [119]. Moreover, cytotoxicity is mediated by the death receptors FAS ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL) [121]. A marker of this degranulation is cell surface expression of lysosomal-associated membrane protein 1 (LAMP1) [122]. Furthermore, NK cells can secrete cytokines, chemokines, and growth factors, such as IFN-γ, IL-13, TNF, FMS-like tyrosine kinase 3 ligand (FLT3L), CC chemokine ligand 3 (CCL3), CCL4, CCL5, and lymphotactin (XCL1) [72,73,123]. NK cells can activate other immune cells following the secretion of these factors. For example, CCL5 and XCL1 attract DCs and FLT3L increases the number of stimulatory DCs in the TME [11,124]. NK cells are required for the anti-tumor CD8+ T cell response by triggering the recruitment of cDC1s and subsequently CD8+ T cells [11,12,125,126,127]. The effects of this cascade are highlighted by patient survival across multiple different cancer types, where the gene signatures of cDC1s, NK cells, and CD8+ T cells all independently predicted improved survival [11,125,128]. Furthermore, IFN-γ production within the TME upregulates MHC-I [129], which causes tumor evasion from NK cells. However, it also results in activated MHC-I presentation of neoantigens for CD8+ T cells. Above all, NK cell-based immunotherapy potentially drives the cancer immunity cycle, indicated by regression and improved patient outcomes.
Unlike T cells, NK cells lack TCRs and do not cause graft-versus-host disease (GVHD) [130,131,132]. NK cell-based immunotherapy presents the possibility of targeting tumors that lack well-defined antigens for specific response and the use of allogeneic products prepared in advance. These facts allow administration in multiple patients without causing GVHD [130,131]. A recent clinical trial demonstrated that ex vivo expanded allogeneic NK cells exhibited enhanced responses against myeloid leukemia. Clinical responses were observed in five of nine evaluable patients, including four complete remissions with low toxicity [133]. Berrien-Elliott et al. reported and summarized the exploration of NK cells as an alternative cell source for allogeneic cell therapies given their inherent ability to recognize cancer, mediate the immune functions of killing and communication, and the fact that they do not induce GVHD, CRS, or immune effector cell-associated neurotoxicity syndrome (ICANS), which indicated low toxicity [134]. Therefore, NK cell-based therapy potentially leads to less toxicity in comparison to CAR-T cell infusions.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24032111
This entry is offline, you can click here to edit this entry!