βA derives from the amyloid precursor protein (APP) [
14] and forms intracerebral senile plaques (SP) that are morphologically heterogeneous and graded according to the Thal scheme [
13]. βA is loosely arranged in diffuse plaques (DP), diffuse amyloid lakes and subpial bands. When harboring thick neurites, SPs are called neuritic plaques (NP) [
15]; these are separately graded by CERAD scheme [
13]. Another NP subtype, the dense-core plaque, displays a central compact βA concretion that is surrounded by a loose zone of diffuse βA deposit [
15]. In EOAD, βA plaques are also often arranged in so-called cotton wool plaques that are large, wispy, spherical structures that lack central βA concretions. Per current neuropathologic scoring schemes, neocortical NP densities are semiquantitatively graded [
13]. The initial stage of βA deposition involves neocortical brain regions (stage I), but with disease progression, there is involvement of allocortical brain (stage II), subcortical nuclei (stage III), medulla oblongata and midbrain (stage IV) and, eventually, pons and cerebellum (stage V) [
13].
In contrast to βA, tau derives from the MAPT gene (chromosome 17). Tau protein is normally found throughout the nervous system and is primarily expressed by neurons. Though its role is not fully elucidated, it is thought to have a role, in part, in neuronal microtubule stabilization. When abnormally hyperphosphorylated, tau is prone to aggregate as intraneuronal paired helical filament inclusions. Within neuronal soma, hyperphosphorylated tau protein forms neurofibrillary tangles (NFT), whereas in distal neuronal processes (i.e., axons and dendrites) it forms neurites and neuropil threads (NT). NFTs primarily localize to limbic regions during early or mild course of disease, but with disease progression the NFTs also aggregate in association cortices and subcortical gray matter nuclei, in a pattern that is distinct from that of βA. A consensus guideline suggests the Braak topographical scheme for NFT staging: stage 0 corresponds to an absence of NFT; stage I/II corresponds to NFT accumulation in transentorhinal regions; stage III/IV corresponds to NFT accumulation within limbic regions; stage V/VI corresponds to wider dispersal of NFT throughout neocortical brain regions [
13].
1.2. Theories on AD Pathogenesis
While the causes of AD-NC are unproven, age is an important risk factor and a number of theories have been posited to explain the gradual age-associated accumulation of βA, NFT and NT inclusions. The
cholinergic hypothesis proposes that AD symptoms arise due to deficiency of intracerebral acetylcholine [
10]. This seminal hypothesis evolved from knowledge that the acetylcholine neurotransmitter and cholinergic neuron loss in limbic and cerebral cortical regions are fundamental features of disease. The
amyloid (or amyloid cascade) hypothesis theorizes that aberrant βA peptides produced by sequential cleavage of APP by APP-cleavage enzymes (i.e., BACE1 and β/γ-secretases) aggregate into oligomers and insoluble extracellular plaques that damage brain neurons and lead to a range of detrimental secondary phenomena [
10,
16,
17]. The
tau propagation hypothesis proposes that development and deposition of aberrant hyperphosphorylated tau protein is the initiating pathological event in AD [
18], whereas the
inflammation hypothesis emphasizes the importance of activated microglia and astrocytes in disease genesis [
10,
19]. Meanwhile, the
oxidative stress hypothesis asserts that a variety of conditions that cause free radical generation lead to peroxidation of membrane polyunsaturated fatty acids, thus precipitating AD lesions [
10]. Similarly, the
mitochondrial hypothesis proposes that mitochondrial dysfunction occurs upstream of the above phenomena, influencing expression and processing of APP and thereby causing aberrant βA accumulation and an associated cascade of secondary brain changes [
10]. The
infectious hypothesis proposes that a pathogen, i.e., virus, bacterium or prion, underpins disease while the
calcium homeostasis hypothesis and
metal ion hypothesis conjecture that calcium or metal ion dysregulation, respectively, are the initial culprit [
10]. The
ion channel [
10],
cell cycle [
20],
autoimmune [
21],
epigenetic [
22] and
granuloma hypotheses [
23], among others, have also been propositioned [
19]. Altogether, the diversity and number of diverging theories for AD highlight the complexity of this neurological condition and the magnitude of uncertainty regarding initial pathogenetic events.
2. Discordance between Neuropathologic Grading Schemes and Theories of AD
Despite the requirement of βA plaques and NFTs for diagnosis of AD-NC and the use of βA as a surrogate endpoint of disease, these markers are of limited utility in disease grading. For instance, precise form(s) and toxic βA species are ambiguous and associations of DP density with cognitive impairment are weak. On the other hand, the abundance and distribution of NFTs and NPs are thought to be predictors for cognitive impairment [13,35]. Yet, NFTs and NPs are also commonly observed in individuals who are cognitively intact [35]. An investigation into risk factors associated with rapid clinical progression, from MCI to dementia, highlights the significance of cerebrospinal fluid (CSF) total tau and phosphorylated tau, but not βA1–42 levels, as pertinent to disease [36]. Moreover, abnormal tau protein inclusions also arise in association with other neurodegenerative conditions (i.e., tauopathies), including Pick disease, progressive supranuclear palsy, corticobasal degeneration, chronic traumatic encephalopathy, aging-related tau astrogliopathy, primary age-related tauopathy, and some forms of frontotemporal dementia [1]. Thus, NFTs and NPs are nonspecific lesions that do not accurately predict elderly persons with AD dementia [37,38]. Meanwhile, a number of alternate brain changes and protein inclusions not encompassed by AD-NC criteria are commonly associated with AD. While AD-NC, vascular diseases, and brain vascular injuries are recognized as the most common neuropathologic lesions associated with AD dementia, these entities also often overlap [39]. For a variety of reasons, it has increasingly been thought that βA is associated with, but not causative of AD. It is also increasingly accepted that significant subsets of individuals with AD dementia exhibit heterogeneous tissue substrates and unique combinations of tissue lesions [40].
Shifting Perspectives: Inconsistencies of the Amyloid Cascade Hypothesis
Despite intensive investigation, direct mechanistic links between βA and AD dementia remain unproven and βA has never been universally accepted as the underlying cause for AD. Over the past decade, a number of studies have suggested that βA toxicity is dependent on the presence of phosphorylated tau inclusions [
18]. Moreover, the ApoE genotype has been recognized to modify the risk of AD and cognitive decline through both βA-dependent and βA-independent mechanisms [
7]. Increasing evidence highlighting discrepancies between AD-NC and established diagnostic criteria for AD dementia has led to reappraisals of the amyloid hypothesis. A revised amyloid hypothesis suggests that lower-order soluble βA polymers that are imperceptible on histology are the true neurotoxic species in AD [
17]. Some researchers maintain that βA and NFTs co-occur, but suggest that disease progression is mediated by tau pathology [
18]. Several investigators now believe that tau pathology is an integral substrate of LOAD and propose that tau toxicity is in some way triggered by βA or βA-related biology. Recent data also uncovers marked variability of tau strains and conformations, and suggests that selective tau recognition by chaperones may differentially influence the accumulation and effects of proteotoxic tau species [
38]. Though tau pathology is largely characterized according to NFT burden, research also highlights a diversity of post-translational tau modifications prior to NFT formation [
44]. As with βA, soluble tau forms are increasingly thought to represent the true neurotoxic species. Some recent data also suggest that AD pathogenesis occurs due to the effects of long-term elevation of βA concentration, rather than by transient overproduction or impaired clearance [
45]. Other studies stress aberrant protein aggregation properties [
46]. Still, epidemiological evidence implies that other phenomena, lesions and/or brain “hits” are contributory [
41,
47,
48] and highlight perplexing questions about AD: why are some mutation carriers from AD kindred resistant to βA plaques and NFTs [
48]? Why does cerebral amyloid angiopathy (CAA) variably co-occur with AD-NC? Why are non-βA and non-tau protein inclusions observed in heterogeneous combination with disease [
40]? Why are vascular pathologies so frequent in association with disease [
41,
42,
43]? Why do SuperAgers exhibit AD-NC, but no clinical evidence of disease [
1,
40]? In light of recent literature, these inconsistencies emphasize the need for distinctions between diagnostic biomarkers and molecular targets for AD therapies [
14].
3. New Theories: Convergence on Vascular and Neuroimmune Homeostatic Factors
Over the past two decades, investigation into alternative and early-stage AD biomarkers has underscored potential new AD targets [17,49]. Given the convergence of a number of early biological phenomena around cerebral vessels, it is now clear that cerebrovascular contributions to AD have been underrecognized. Increasing data show that inflammatory changes and loss of vascular wall integrity are the earliest identifiable lesions in persons with AD, preceding βA and tau accumulation [50,51].
In parallel, improvements in microscopy and imaging technologies have led to revised concepts in neuroanatomy, neuroimmunology, and brain barrier biology. The meningeal lymphatic system has emerged as a critical player in neurophysiology [57,58,59].
3.1. Brain Border Macrophages in AD
Pivotal roles of macrophages at brain surfaces and their significance in brain homeostasis are highlighted in recent literature [
66,
67,
68,
69,
70]. Within the cranium, but outside brain parenchyma, macrophages known as central nervous system (CNS)-associated macrophages (CAMs) or parenchymal border macrophages (PBMs) reside within meninges, PVS, and choroid plexus and are involved in regulating essential exchange between CNS parenchyma and the periphery. These macrophages are thought to support and maintain brain barrier properties, control drainage of CNS antigens, and aid in clearance of waste metabolites [
66,
67]. Moreover, macrophage populations at brain borders are shown to be phenotypically diverse [
66,
68]. Due to their strategical positioning as well as roles in CSF processing and coordination of reciprocal cross-talk with other cell types, CAMs/PBMs act as critical regulators of brain metabolism and gatekeepers of general neuroimmune processing [
67,
68]. Evidence suggests that they control the environment and tune entry of immune cells from the CSF and blood into the brain parenchyma and, in turn, regulate the exchange of diverse molecules between the bloodstream, periphery, and brain [
67]. PVS macrophages are increasingly thought to have underrecognized roles in CAA and AD [
68,
69]. In a transgenic TgCRND8 AD mouse model, their depletion by clodronate liposome resulted in a prominent increase in PVS βA
1–42 deposits [
70].
3.2. Peripheral Monocytes and Macrophage Infiltration in AD
While microglia are the principal innate immune cell of the brain and originate from yolk sac erythromyeloid progenitor cells from which they migrate, propagate, and spread during embyogenesis, it has been shown that peripheral monocytes are continuously replenished in adult brains [
71,
72]. This includes healthy adult brains, in which the recruited cells engraft and differentiate into parenchymal microglia, i.e., new brain resident cells, or remain a distinct population [
73,
74,
75]. While this recruitment occurs in limited numbers in healthy subjects, experimental AD subjects exhibit accelerated recruitment of monocytes from the periphery [
73,
74,
75]. Invading monocytes and macrophages that engraft the brain have been shown to derive from circulating Ly-6C
hi monocytes or from bone marrow-derived progenitors such as granulocyte–macrophage progenitors (GMPs), or other hematopoietic stem cell progeny [
66,
73,
74].
3.3. Microgliosis and Other Vascular-Immune Factors in AD
Reactive microglia, i.e., resident brain phagocytes, invariably accompany AD-NC [
76,
77,
78,
79,
80,
81,
82,
83]. In fact, activated microglia and βA deposits spatially overlap in cerebral cortices of subjects with MCI [
77,
82], while baseline brain microglial activation and activated hippocampal microglia are significantly increased in subjects with AD [
71]. In response to the accrual of βA plaques, adjacent microglia increase their expression of CD11b, CD68, and complement receptor 3 [
82,
84]. Moreover, in vitro analyses demonstrate the ability of βA to stimulate pro-inflammatory cytokine (including interleukin (IL)-1β, IL-6, IL-8, tumor necrosis factor (TNF)), chemokine, and neurotoxin production by human microglia, causing neuronal damage [
85,
86,
87]. In response to βA, microglia also secrete proteolytic enzymes and express receptors to promote βA phagocytosis and clearance. This includes the upregulation of class A scavenger receptors (SR-A), the receptor for advanced glycation end-products (RAGE), CD36, toll-like receptors (TLRs) and b1 integrins [
82,
88]. Therefore, βA may signal in combination with a host of immune factors to facilitate the recruitment of peripheral immune cells into brains of persons with AD, which may impact on brain function [
75].
TREM2 (triggering receptor expressed in myeloid cells 2) has also emerged as a key player in microglial and AD biology [
71,
89,
90]. This protein has been shown to function as a receptor for βA and has affinity for its oligomeric forms [
89]. The TREM2 cell surface receptor promotes microglial association with βA plaques and via its interaction with the activating adaptor protein DAP12, it plays critical roles in chemotaxis, survival, and proliferation of myeloid cell populations and phagocytosis of a variety of substrates, including apoptotic neurons, bacteria, low-density lipoprotein (LDL) and other lipoproteins, and βA [
71]. Loss of TREM2 function diminishes microglial responses to βA plaques, enabling a more toxic state [
90].
3.4. Imaging Evidence of Early Hypometabolism and Vascular and Perivascular Dysfunction
In support of a vascular–immune theory of AD, clinical imaging evidence demonstrates that subtle, asymptomatic vascular changes and blood flow aberrations precede βA and NFT formation, cerebral cortical atrophy, and cognitive decline [
51,
96,
97,
98]. 18F-FDG-PET imaging studies document decreased glucose uptake in specific brain regions during prodromal stages of AD [
99]. As abnormal FDG uptake is an indicator of resting-state brain glucose hypometabolism, this represents a biomarker of brain vascular dysfunction. Longitudinal 18F-FDG-PET studies have shown that reduced hippocampal FDG uptake predicts cognitive decline with high sensitivity and specificity years in advance of clinical AD symptoms [
100]. Furthermore, high resolution dynamic contrast-enhanced (DCE) MRI analyses demonstrate blood–brain barrier (BBB) injury in the hippocampi of individuals with MCI, suggesting vascular tunica intima damage as an early independent biomarker of cognitive impairment [
50,
51]. DCE MRI analyses also suggest that BBB breakdown contributes to ApoE4-associated cognitive decline, independent of AD-NC [
98].
3.5. Cardiac and Cardiovascular Disease Effects in AD
Clinicopathologic investigations document a variety of cardiac and extracranial and intracranial vascular changes and pathologies in persons with AD [
1,
106,
107,
108,
109,
110,
111,
112,
113,
114]. Cardiac and extracranial cardiovascular diseases associated with AD include structural or functional heart diseases such arrhythmic disorders and coronary artery, valvular, and hypertensive diseases [
1,
112]. Associated extracranial carotid artery changes include stenosis, occlusion, and emboli, whereas intracranial cerebral large vessel pathologies include circle of Willis atherosclerosis, vasculitis and vasculopathies [
1,
112]. Intracerebral small vessel diseases encompass a multitude of processes, most notably CAA and arteriolosclerosis [
1,
112].
Microscopic and macroscopic infarcts, i.e., secondary effects of cardiovascular diseases, are predictive of brain neuron and axon density as well as AD dementia risk [
1,
106]. Longitudinal community-based studies confirm that macroscopic and microscopic infarcts are independent predictors for cognitive decline, though pathophysiological mechanisms are not fully defined [
104,
107,
108]. Other cardiovascular risk factors such as atrial fibrillation and congestive heart failure are also linked to frequency of AD-NC [
105,
106,
108].
4. Potential Roles of the Glymphatic–Lymphatic System in AD
4.1. Clearance Mechanisms at the Neurovascular-Perivascular Interface
Aside from permitting nutrient transfer into brain, the neurovascular–perivascular unit (NVU/PVU) is a critical brain border and vital interface responsible for the transfer of metabolic wastes, immune cells, immune signals, and other small molecules into and out of brain [
102,
104]. Within the brain, the NVU is comprised of endothelial cells, pericytes, smooth muscle cells, reticular cells, macrophages, and astrocytes, among other cell types, whereas PVUs are situated along the perimeter of intracerebral blood vasculature (
Figure 1). Though cellular and molecular mechanisms involving the NVU/PVU remain under investigation, knowledge on this brain barrier is evolving swiftly and available data suggest that anatomic, physiologic and pathologic changes at this interface are essential to brain homeostasis [
69,
104]. Brain metabolites, including βA, are cleared from the brain by ISF, CSF and blood [
125]; through phagocytosis by pericytes, vascular smooth muscle cells, macrophages, and glial cells; through transcytotic processes across the BBB; and through receptor-mediated endocytosis involving various cell types [
14,
126,
127] (
Figure 2). Detection of tau and other brain proteins within blood implies that they are also cleared from the brain to the periphery through the NVU/PVU [
125,
128]. In combination with preclinical evidence on glymphatic–lymphatic functions [
58,
59,
62], knowledge on NVU/PVU biology highlights the potential significance of cerebral vascular and perivascular tissue change and pathology in AD, as NVU/PVU alterations are likely associated with impaired brain metabolite clearance and hampered brain–brain and brain–periphery signaling, in addition to inefficient brain nutrient delivery [
129,
130,
131]. Thus, NVU/PVU dysfunction may have regional and/or global brain effects, and may lead to generalized protein clearance failure and dysregulated brain–body communication.
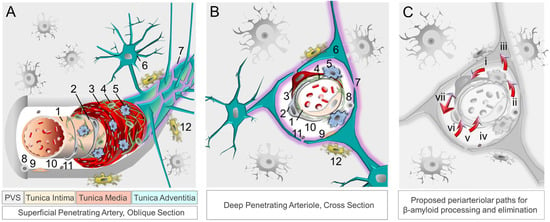
Figure 1. The neurovascular–perivascular unit and recognized βA and brain metabolite clearance mechanisms. Neurovascular and perivascular elements facilitate nutrient exchange as well as metabolism and removal of metabolic wastes, including βA. The neurovascular unit, depicted in oblique section (
A) and cross section (
B), is composed of (1) endothelial cells, (2) pericytes, (3) smooth muscle cells, (4) reticular cells, (5) macrophages, and (6) astrocytes comprised of (7) aquaporin-4 expressing astrocytic end-feet. The perivascular space harbors (8) other immune cells, (9) fibroblasts, (10) apolipoprotein E, (11) and extracellular matrix components, while (12) microglia are present in adjacent brain tissue. Metabolites, including βA and tau, are ultimately cleared from the brain by interstitial fluid flow in combination with other mechanisms (
C), including phagocytosis [by (i) vascular smooth muscle cells, (ii) macrophages, (iii) glial cells, and (vi) pericytes]; (iv) apolipoprotein E-mediated processes); (v) transcytosis across the blood–brain barrier; receptor-mediated endocytosis (involving various immune and other cell types); (vii) perivascular glymphatic mechanisms [
62,
63,
121]. Abbreviation: β, beta; PVS, perivascular space.
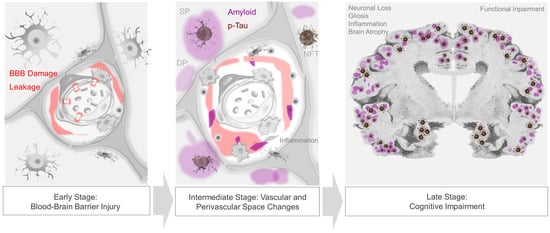
Figure 2. Progression of neurovascular–perivascular and parenchymal brain changes in AD. Blood–brain barrier (BBB) damage and infiltration of serum and blood cellular elements occur in prodromal, i.e., early-stage AD (left panel). Vascular and/or inflammatory insults may further induce transudates in perivascular spaces, impede interstitial fluid drainage, and cause impaired nutrient exchange, edema, cytokine and metabolic waste accumulation, and reactive inflammation and dysregulation (middle panel). In later stages of disease, extracellular βA plaques and intraneuronal neurofibrillary tangles, as well as other protein inclusions, are frequent and are associated with progressive cognitive impairment (right panel).
4.2. Compound Proteinopathies in AD
As highlighted in recent literature, significant subsets of AD dementia cases are attributable to TDP-43 inclusions and/or α-synuclein-positive Lewy bodies [1,132,133]. Studies show that non-βA and non-tau proteinopathies are not mutually exclusive of one another, nor with AD-NC [132,133]. In fact, cohort studies demonstrate that TDP-43 pathology combined with AD-NC accounts for 35–37% of pathology in elderly subjects with AD dementia [134,135], while Lewy body inclusions overall manifest in more than 50% [135,136]. Thus, common mechanisms may exist for proteinopathic diseases [1,137].
4.3. Blood, Peripheral Signaling and Brain-Body Connections in AD
Intracranial vessels (i.e., lymphatic and blood vascular) and biofluids (i.e., ISF, CSF, and blood) convey innumerable signals. Secretory mechanisms at the BBB impart endocrine-like properties of BBB endothelium [138,139]. Moreover, neurons and neuroglial cells communicate with themselves, with one another, and with the periphery via receptors and extracellular molecules involving autocrine, paracrine, and endocrine signals that transmit in extracellular spaces [138,139,140]. Other serum-derived factors and biomolecules derived from brain and periphery are also transported through intracranial fluids [141,142,143,144,145,146,147].