Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Trientine, a copper-chelating drug used in the management of patients with Wilson’s disease, demonstrates beneficial effects in patients with hypertrophic cardiomyopathy.
- cardiomyopathy
- diabetes
- hypertensive
- TETA
1. Introduction
Heart failure affects more than 64 million people globally [1], with its prevalence ranging from 0.12 to 6.70% [2]. It is a complication of other diseases such as diabetes mellitus [3], ischemic heart disease, or long-standing hypertension [4], or primarily due to genetic and idiopathic conditions [5]. These conditions can give rise to the development of cardiomyopathy. Myocardial hypertrophy is the most common cause of heart failure [6], characterized by enlargement of the heart [3].
Myocardial hypertrophy occurs following remodeling of the heart, triggered by various stimuli. Remodeling causes myocardial structural, molecular, and cellular changes, leading to alterations in cardiac function, size, and shape [7]. Many pathological events, such as neurohormonal activation, cardiac volume overload, and pressure overload are thought to be centrally involved in the development of cardiac hypertrophy [7]. Deficiencies in micronutrients such as copper, zinc, and selenium may also play a role in the development of heart failure [8]. Abnormality in copper homeostasis manifested by a deficiency in myocardial copper may contribute to the pathogenesis of cardiac hypertrophy [9]. Patients with ischemic heart disease tend to have a lower cardiac copper content [10], possibly due to increased myocardial copper efflux [11] and reduced activity in certain copper-dependent enzymes [10]. Animals fed a diet deficient in copper had significantly heavier hearts [12], owing to alterations in heart biochemical properties and morphology [13]. Similar observations were also noted in pressure-overloadinduced hypertrophied hearts in rats [14,15].
The exact mechanism of how copper deficiency induces cardiac hypertrophy is not well understood but may be partly attributable to dietary and hereditary etiologies. Genetic polymorphism in copper-transporting ATPase has been postulated to cause pathological cardiac changes, as observed in Menkes’ disease, an X-linked genetic defect affecting energy-dependent copper transporters [16]. Copper plays an important role in many cellular processes such as antioxidant activity and mitochondrial respiration [17]. Dietary supplementation of copper reversed cardiac hypertrophy in rats [18].
Cardiac hypertrophy and heart failure are managed pharmacologically using angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, calcium channel blockers [19], β-blockers, or angiotensin receptor–neprilysin inhibitors [20,21,22]. Due to the discovery of copper-deficiency-induced cardiac hypertrophy, clinical trials have been conducted to investigate the effects of trientine, a copper chelator, on cardiac hypertrophy or hypertrophic cardiomyopathy [23,24].
Trientine, also known as triethylenetetramine (TETA) dihydrochloride (Figure 1), is an orphan drug [25]. It is an organic compound with a molecular formula of C6H18N4, which was originally approved in 1985 by the U.S. Food and Drug Administration as second-line therapy for patients with Wilson’s disease who cannot tolerate penicillamine [26,27]. Wilson’s disease is a genetic condition that arises from copper accumulation in the body, particularly the liver. This could lead to liver cirrhosis and degenerative neurological conditions which could be fatal [28]. Trientine chelates hepatic excess copper and increases urinary copper excretion, leading to hepatic improvement in the majority of patients with the disease. Other copper modulators are dimercaprol and zinc salts (acetate, sulfate, and gluconate) [27].
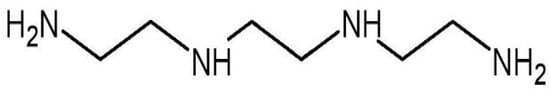
Figure 1. Molecular structure of trientine.
The role of trientine in cardiomyopathy has gained much interest, although its mechanistic action is poorly understood.
2. Role of Trientine in Cardiac Copper Regulation
Copper is required for the normal function and structure of the heart. It is involved in various cellular metabolic activities such as iron and zinc uptake, as well as oxyradical scavenging [17]. It enters cells via plasma membrane copper transporter-1 (CTR-1) and copper transporter-2 (CTR-2) [29]. Once inside the cells, the ion is delivered to its targets by various chaperones [17]. However, in excess, it may be detrimental due to the generation of reactive oxygen species (ROS). Consequently, its uptake, distribution, and elimination are tightly governed to maintain cellular homeostasis [29,30].
The serum copper level is higher in patients with cardiomyopathy compared with that of normal individuals [31]. It is postulated that increased efflux of copper from the myocardial cells leads to a high level of serum copper [32]. To confirm this, many animal model experiments using various cardiomyopathy models such as pressure-overload-induced cardiac hypertrophy [14] or diabetic cardiomyopathy [33] have been conducted. Findings from the studies confirmed the depletion of myocardial copper content measured in left ventricles [14,15,33]. The depleted content could not be replenished even in the presence of higher circulatory copper levels [11]—a phenomenon which remains elusive.
Copper exists in two valencies: Cu(I) and Cu(II). Cu(I) is mainly present intracellularly and contributes 95% of total body copper, and the remainder exists as Cu(II) in the extracellular space [34]. Its transport intracellularly is governed by various chaperones which will be discussed in the subsequent subtopics. Trientine selectively forms a complex with Cu(II), not Cu(I) [35], and serves as a copper chaperone to transport the ion to other copper-binding molecules in copper-depleted cardiomyocytes [14]. However, the mechanism is still unclear. Cu(II)-trientine enters cells as an intact complex via active transport [11]. CTR-1 expression is the major copper transporter in the heart. However, trientine does not affect myocardial CTR-1 expression [14]. Even in diabetic myocardium, it does not restore the diminished CTR-1 protein expression [34]. In cardiomyocytes transfected with CTR-1 gene silencing using siRNA, trientine-facilitated copper uptake into cardiomyocytes was unaffected, observed by increased copper accumulation in the cells. This was different from the cells that were exposed to Cu(II) chloride only, which demonstrated a decline in cellular copper content [11], confirming that the influx of copper into cells by trientine is CTR-1-independent. Studies investigating the effects of trientine on CTR-2 reported inconsistent findings (Table 1). Trientine exhibited elevated CTR-2 expression in a few studies [15,34] but no effect in another study [14]. This suggests that CTR-2 may partly contribute to copper transportation into cardiomyocytes. CTR-2 expression in the heart is determined by the cellular copper status, which is reduced in copper deficiency [36]. Therefore, trientine may upregulate CTR-2 expression indirectly due to the accumulated copper in cardiomyocytes. Another transporter that may be involved in copper transport is chloride channel CLC17 [37], and the effects of trientine on the transporter should be investigated. More studies need to be conducted to better understand the mechanisms of copper uptake by the drug.
Further studies in animals demonstrated that trientine restored myocardial copper content in cardiomyopathy models [14,34,35] (Table 1). It was previously revealed that trientine at a low dose (21.9 mg/kg, orally twice daily for 6 weeks) replenished deprived copper in the heart, but a relatively high dose (87.6 mg/kg, orally twice daily), an equivalent dose commonly used for the treatment of Wilson’s disease, failed to reload the loss of myocardial copper content in rats with pressure-overload-induced cardiac hypertrophy. Moreover, the high dose of trientine decreased copper content in the heart of normal rats, whereas the same phenomenon was not observed with the low dose [14]. The findings suggest that trientine at a relatively high dose of more than 175 mg/kg/day promotes the removal of copper from the heart. The protective effect of trientine on the myocardial copper content agreed with other animal studies that adopted a trientine dosage of approximately 100 mg/kg/day or less [15,33,34,35,38,39].
Administration of trientine exhibited protection against left ventricular hypertrophy in diabetic patients [40]. Clinical trials reported that serum copper was unaltered by trientine therapy after six [23] or twelve months [40] in patients with hypertrophic cardiomyopathy. Yet, the 24 h urinary copper level was significantly elevated in the patients, indicating increased excretion of excess copper (Table 1). However, a small significant rise in serum ceruloplasmin, the main copper-bearing protein in the blood, occurred in the patients, indicating increased cellular uptake of copper [23].
Despite the effects of trientine on myocardial copper content, it has no significant effects on plasma copper levels in rats, even though it reduces renal copper content and facilitates urinary excretion of excessive copper in the blood [14,35,39]. In other words, trientine therapy is not likely to cause systemic copper deficiency. A similar insignificant effect on plasma copper was also observed in patients with left ventricular hypertrophy who were receiving trientine. The effectiveness of the therapy was monitored by a decrease in left ventricular mass [40].
Regardless of its beneficial effects on diabetes-induced copper depletion in the heart [34,39], trientine has no significant effect on blood glucose levels in diabetic rats [25,34,39,41,42]. How it offers cardioprotection without affecting blood glucose is not understood. More mechanistic studies in experimental animals should be conducted to elucidate the mechanism.
Table 1. The effects of trientine on cardiac copper regulation in animals and related blood parameters in animals and patients with hypertrophy.
| Study | Type of Model/Subjects | Trientine (TETA) (Dose and Duration) |
Findings | Reference |
|---|---|---|---|---|
| Animal | STZ-induced diabetic cardiomyopathy in rats |
20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ cardiac copper content ↔ CTR-1 mRNA and protein ↑ CTR-2 mRNA and protein |
[34] |
| Animal | STZ-induced diabetic cardiomyopathy in rats |
20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ cardiac copper content | [38] |
| Animal | Transverse aortic constriction- induced cardiac hypertrophy in rats |
21.9 and 87.6 mg/kg twice daily orally for 6 weeks | TETA (21.9 mg/kg/day): ↑ LV copper content ↑ urinary copper ↓ renal copper content ↔ plasma copper level ↔ CTR-1 and CTR-2 protein TETA (87.6 mg/kg/day): ↓ LV and renal content ↑ urinary copper ↓ renal copper content ↔ plasma copper level ↔ CTR-1 and CTR-2 protein |
[14] |
| Animal | Ascending aortic constriction-induced cardiac hypertrophy in rats | 21.9 mg/kg twice daily orally for 6 weeks | ↑ cardiac copper ↔ CTR-1 mRNA and protein ↑ CTR-2 mRNA and protein |
[15] |
| Animal | STZ-induced diabetic cardiomyopathy in rats |
(a) Intravenous infusion, 60 s once hourly in increasing doses (0.1, 1.0, 10, and 100 mg/kg) (b) 8–11 mg/day in drinking water for 7 weeks (post-treatment) |
↑ Copper urinary excretion ↑ Cardiac total copper |
[39] |
| Animal | STZ-induced diabetic cardiomyopathy in rats |
30 mg/day in drinking water for 8 weeks (post-treatment) | ↑ LV total copper | [33] |
| Human | Type 2 diabetic patients with LVH (n = 15) | 600 mg twice daily orally for 12 months | ↔ plasma copper ↑ 24 h urinary copper |
[40] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg twice daily if tolerated for 6 months | ↔ serum copper | [23] |
CTR-1, copper transporter 1; CTR-2, copper transporter 2; LV, left ventricle; LVH, left ventricular hypertrophy; STZ, streptozotocin; ↑, significant increase; ↓, significant decrease; ↔, no difference.
3. Effects of Trientine on Mitochondrial Function and Biogenesis
Copper is an essential trace element that serves as a co-factor in various enzyme activities involved in mitochondrial ATP production [8,43]. Copper is delivered to its target molecules in various organelles by chaperones [8]. Upon entering cells, copper is fetched by cytosolic copper chaperones. Cytosolic soluble cytochrome c oxidase (Cco) copper chaperones 17 (Cox17) and 11 (Cox11) are chaperones that shuttle copper to other chaperones such as mitochondrial-inner-membrane-bound Cco-assembly proteins—Sco1 and Sco2—which then deliver the copper to Cco in the mitochondria [37,44]. Cco is a copper-dependent enzyme that is actively involved in oxidative phosphorylation in the electron transport chain [44]. In cardiomyopathy, its activity is reduced [38,45].
Only two studies have investigated the effects of trientine on myocardial mitochondrial function and biogenesis. Zhang et al. [38] demonstrated that myocardial copper replenishment by trientine in the copper-deficient myocytes of diabetic rats restored normal copper trafficking between the cytoplasm and mitochondria, as evidenced by an increase in gene expressions of copper chaperones Cox17, Cox11, and Sco1 (Table 2) (Figure 2). The drug increased Cco subunits I (mt-coI) and II (mt-coII) gene expression but did not affect subunit III (mt-coIII). However, trientine did not alter the protein expression of the subunits [38]. Future studies should be conducted to investigate the effects of the drug on the mitochondrial membrane potential in cardiomyopathy since Cco and Cox17 directly affect the potential. The mitochondrial membrane potential is a crucial entity in energy storage during oxidative phosphorylation. Disturbance in the potential generation could result in mitochondrial dysfunction [46].
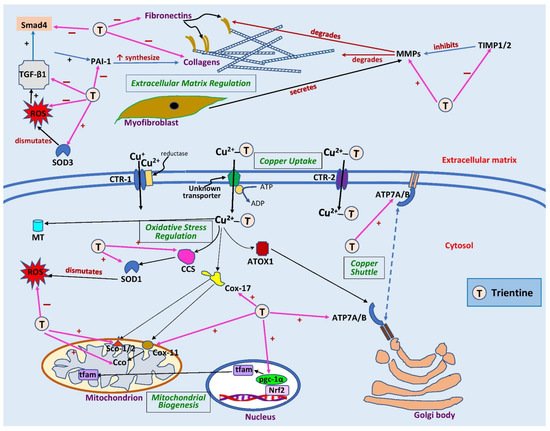
Figure 2. Sites of action of trientine in cardiac remodeling. Extracellularly, trientine reduces interstitial fibrosis by inhibiting tissue inhibitor of metalloproteinase 1/2 (TIMP1/2), leading to an increase in matrix metalloproteinases (MMPs). This, in turn, augments collagen degradation. Inhibitory effects of trientine on transforming growth factor-β1 (TGF-β1), plasminogen activator inhibitor-1 (PAI-1), and small mothers against decapentaplegic (Smad4) lead to decreases in expression of matrix proteins (collagen and fibronectin). The complex of trientine-Cu(II) is brought into cells by an unknown transporter and partly by copper transporter-2 (CTR-2). The drug increases the expression of superoxide dismutase-1 (SOD1) and -3 (SOD3) as well as copper chaperone for superoxide dismutase-1 (CCS), leading to a decrease in reactive oxygen species (ROS) formation. Trientine also elevates the expression of cytosolic soluble cytochrome c oxidase (Cco) copper chaperones 11 (Cox11) and 17 (Cox17), cytochrome c oxidase assembly protein-1/2 (Sco1/2), and peroxisome proliferator-activated receptor coactivator-1α (pgc-1α), which improves mitochondrial biogenesis and function. It also augments the expression of ATPase copper-transporting α (ATP7A) but has no effect on ATPase copper-transporting β (ATP7B) or antioxidant 1 copper chaperone (ATOX1), which are responsible for shuttling copper into trans-Golgi network or secretory vesicles. CTR-1, copper transporter 1; MT, metallothionein; nrf2, nuclear factor erythroid 2-related factor 2; tfam, mitochondrial transcription factor A; ↑, significant increase; +, stimulates; −, inhibits. Blank arrows demonstrate subsequent events; brown arrow indicates degradation, dashed blue arrow displays translocation of copper, and pink arrows indicate the sites of action of trientine.
Table 2. Effects of trientine on copper chaperones involved in cardiac mitochondrial function and biogenesis in human and animal studies.
| Study | Type of Model/Subjects | Trientine (Dose and Duration) |
Findings | Reference |
|---|---|---|---|---|
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ ATP7A ↑ Atp7a mRNA |
[34] |
| Animal | STZ-induced diabetic cardiomyopathy in rats | 20 mg/day in drinking water for 8 weeks (post-treatment) (~68 mg/kg/day) | ↑ mt and cyto cox17 mRNA and protein ↑ mt and cyto cox11 protein ↑ mt Sco1 protein in LV ↑ mitochondrial Cco activity ↑ mt-coI mRNA ↑ mt-coII mRNA ↔ mt-coIII mRNA ↔ mt-coI protein ↔ mt-coII protein ↔ mt-coIII protein ↔ mt-DNA content ↔ mt-tfam mRNA ↔ mt-ssbp mRNA ↑ pgc-1α mRNA |
[38] |
| Human | Patients with hypertrophic cardiomyopathy (n = 20) | 300 mg twice daily orally, increased after 1 week to 600 mg twice daily if tolerated for 6 months | ↔ PCr/ATP ratio | [23] |
ATP7A or Atp7a, ATPase copper-transporting α; cox11, cytosolic soluble cytochrome c oxidase (Cco) copper chaperone 11; cox17, Cco copper chaperone 17; cyto, ctytosol; LV, left ventricular; mt, mitochondria; mt-coI, mitochondrial cytosolic soluble cytochrome c oxidase (Cco) subunit I; mt-coII, mitochondrial Cco subunit II; mt-coIII, mitochondrial Cco subunit III; mt-tfam, mitochondrial transcription factor A; mt-ssbp, mitochondrial single-strand DNA-binding protein; PCr, phosphocreatine; pgc-1α, peroxisome proliferator-activated receptor coactivator-1α; Sco1, cytochrome c oxidase assembly protein-1; STZ, streptozotocin; ↑, significant increase; ↓, significant decrease; ↔, no difference.
Trientine also enhanced the expression of ATPase copper-transporting α (ATP7A) localization and its gene expression (Atp7a) in left ventricular diabetic rats [34]. ATP7A is a copper chaperone that delivers copper to ceruloplasmin in the Golgi bodies [37,44]. In terms of mitochondrial DNA synthesis, trientine did not influence mitochondrial transcription factor A (mt-tfam) or single-strand DNA-binding protein (mt-ssbp) gene expression, leading to no change in mitochondrial DNA content. However, it augmented the expression of peroxisome proliferator-activated receptor coactivator-1α (pgc-1α), a principal regulator of mitochondrial metabolism [38].
The heart has a high energy metabolism, hence it requires an energy depot for an immediate supply of ATP. Phosphocreatine represents energy storage for the rapid production of ATP. The former converts to creatine, releasing its phosphate that phosphorylates ADP, synthesizing ATP [47]. Hence, the ratio could serve as an indicator of cardiac high-energy phosphate metabolism [48]. This can be measured by using phosphorus magnetic resonance spectroscopy [49]. In patients with abnormalities in cardiac metabolism, the myocardial phosphocreatine/ATP ratio declines, and this has been shown to be prognostically relevant in models of heart failure [48]. Only one clinical trial in phase 2a has explored the effects of trientine on cardiac energetics in 20 patients with hypertrophic cardiomyopathy [23]. They found that trientine therapy at 600 mg twice daily for six months produced a non-significant 10% rise in the left ventricular phosphocreatine/ATP ratio, suggesting the potential of trientine to restore mitochondrial function and energetics in cardiomyopathy.
Collectively, trientine therapy can reverse cardiomyocyte-defective copper metabolism in cardiac hypertrophy or cardiomyopathy. This could improve mitochondrial function and bioenergetics, driving amelioration of heart structure and function. More studies on animals investigating this aspect should be conducted. Trientine may affect sirtuin-3, which is crucially involved in mitochondrial metabolism. It warrants further investigation to appreciate its role in mitochondrial function and biogenesis.
This entry is adapted from the peer-reviewed paper 10.3390/ph15091145
This entry is offline, you can click here to edit this entry!