Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Medicinal
The pyrazole moiety is a heterocyclic ring system (five membered) with 3 C and 2 N in adjacent sites. Pyrazole compounds have an extensive past of applications, being used as herbicides, agrochemicals, and as active pharmaceutical agents. Pyrazole derivatives exhibit anti-inflammatory properties and have been shown to target several cancer cell lines, such as COX (I/II).
- pyrazole
- biomolecules
- anticancer
- anti-inflammatory
1. Introduction
Cancer is an uncontrollable abnormal cell proliferation that is a severe life-threatening disorder worldwide. Pharmaceutical firms are now investing heavily in developing and enhancing viable and safe anticancer treatments with minimal side effects. Pyrazole derivatives exhibit anti-inflammatory properties and have been shown to target several cancer cell lines, such as COX (I/II). The description includes a library of powerful compounds against cancer cell lines and inflammation. Among them, certain effective compounds and their SAR structures are strongly associated with biological activity and predicted some additional derivatives that approach structural alterations. Researchers addressed the anticancer therapeutic action of compounds; all derivatives showed strong inhibitory activity, but certain powerful compounds demonstrated good inhibitory activity against cancer cell lines.
2. Pyrazole Biomolecules as Cancer Therapeutics
Compound 21 exhibited the most strong biological activity against HCT116, MCF-7, and Aurora-A kinase inhibitory activity cell lines, with IC50 values of 0.39 ± 0.06 µM, 0.46 ± 0.04 µM, and 0.16 ± 0.03 µM, which were equivalent to the positive control. The scaffolds of the known inhibitors of Aurora-A kinase can be categorized into four main groups, A–D. The scaffold in A contains 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole; the scaffold in B contains pyrrolo[2,3-b]pyrimidine; the scaffold in C contains quinoline; and the scaffold in D contains 2-anilino-diaminopyrimidine. Benzamide is a significant pharmacophore of natural compounds as well as a synthetic precursor for a variety of medications. Benzamide has been shown to have several pharmacological effects, including anti-inflammatory, anticancer, and anti-fungal properties. To create a more potent reaction with the Aurora-A kinase domains, a benzamide moiety was transferred to the pyrazole moiety to create the N phenyl-1H-pyrazole-4-carboxamide template. The highest activity was found for 21 compounds with a para-NO2 group on the A-rings and a para-OEt group on the B-rings [44] (refer to Figure 4 for Structure and SAR). In a separate investigation, compound 30 demonstrated a significant inhibitory effect against WM266.4 and A375 with IC50 values ranging from 1.50 to 1.32 µM. It makes use of a common scaffold comprised of a terminal aromatic group (phenyl or pyrazole) that occupies the allosteric pocket generated by the displacement of the DFG loop and an amide linker that links an aryl group in the hydrophobic pocket to a hinge-binding heterocycle. The use of an amide bond would allow for the quick and efficient screening of several candidate hinge-binding groups. The original molecule was modified to see how the substituents affected the BRAFV600E inhibitory effects. Urea derivatives have higher inhibitory effects than Schiff base compounds and related intermediate amines. Changes in substituents on the benzene rings had a minor impact on the activity of the compounds. In contrast, differences in the skeleton had a greater impact, which may explain why substituents on the benzene rings had a minor impact on the ability for molecular bonding between BRAFV600E and the compounds. According to the findings, the pyrazolyl core and the phenyl urea motif are critical in the inhibition of BRAFV600E [51] (refer to Figure 4 for Structure and SAR).
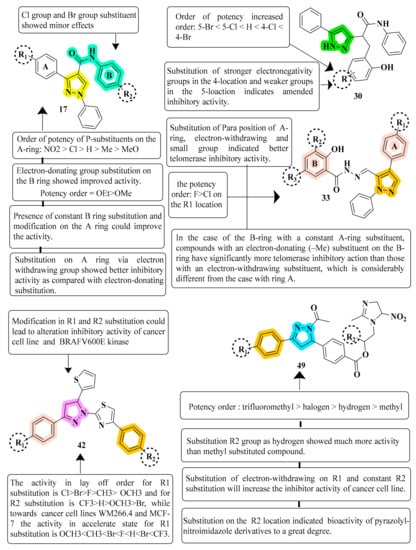
Figure 4. Structure and SAR of potent anticancer activity compounds 17, 30, 33, 42, and 49.
Compounds with the OH and Me groups on the B-ring, compound 33, resulted in notable activity. Compound 33 displayed the most powerful effect, inhibiting the growth of MCF-7 and B16-F10 cells with IC50 values of 0.57 ± 0.03 and 0.49 ± 0.07 µM, respectively, and inhibiting the activity of telomerase with an IC50 of 1.9 ± 0.43 µM. More than one way to achieve powerful coordination may be achieved by using hydrazone derivatives and, specifically, acyl hydrazones. Acylhydrazone derivatives have the potential to form complexes with metal ions, such as Fe3+ and Ni2+, resulting in the inhibition of a physiologic reaction catalyzed by ions or the promotion of the absorption, transportation, distribution, and metabolism of drugs in the body, respectively. The F group outperformed the Cl group in antiproliferative action on the A ring, which might be attributable to the fluorine substituent’s greater lipophilicity and metabolic stability. The presence of fluorine often increases the solubility of lipids, which increases the rates of absorption and transport of drugs. Compound 33, based on its data structure, may be useful in designing and manufacturing stronger telomerase inhibitors [54] (refer to Figure 4 for Structure and SAR).
A series of novel BRAF-targeting pyrazole-based derivatives developed recently may prove useful against cancer. Some of them proved to have potential antitumor activity, particularly compound 42, which had an IC50 value of 0.12 µM against the cell line WM266.4 and 0.16 µM against the cell line MCF-7 and which also showed the most potential antiproliferative activity of apoptosis by flow cytometry experiments. Common scaffolds consist of a terminal aromatic group (phenyl or pyrazole) that fills the allosteric pocket. Thriophene and thiazole groups were demonstrated to have antibacterial, anti-inflammatory, and antiviral properties according to generalized research. The relevant compounds were formed by displacing the DFG loop and an amide linker and linking an aryl group in the hydrophobic pocket to a hinge-binding heterocycle. The introduction of an amide bond would enable the rapid and effective screening of different hinge-binding groups. Changes in substituents on the benzene thiazole rings had only a small effect on the compounds’ activity. As a consequence, compound 42 and the other pyrazole derivatives with effective thiazole and thiophene pharmacophores are promising candidates for further research as anticancer medicines [62](refer to Figure 4 for Structure and SAR).As shown in the figure below, compound 49 acts as a potent inhibitor of HER-2/EGFR, possessing a half-life of 0.26 m/0.51 m compared to the positive controls erlotinib (IC50 = 0.41 µM for HER-2 and IC50 = 0.20 µM for EGFR) and lapatinib (IC50 = 0.54 µM for HER-2 and IC50 = 0.28 µM for EGFR). On the mechanistic level, nitroimidazole derivatives drew much attention because of their ability to enter and accumulate in tumor areas. To boost the antiproliferation activity of pyrazolyl-nitroimidazole derivatives against Hela or HepG2 cell lines, electron-withdrawing groups were shown to be preferred to electron-donating groups. This indicates that the synthesized chemicals’ significant inhibitory effects on cell growth were directly connected to their kinase inhibitory actions [68] (refer to Figure 4 for Structure and SAR).
These molecules are intriguing novel frameworks for the case of cancer therapies due to their simplicity of synthesis and extraordinary biological activity. Compound 50 inhibited MCF-7, A549, and HeLa cells significantly, with IC50 values ranging from 0.83–1.81 µM. Furthermore, the compounds induced G1 phase cell cycle arrest in MCF-7 cells by inhibiting cyclin D1 and CDK2. It was observed that structural alterations and modifications on the B ring of pyrazolobenzimidazole derivatives resulted in distinct cytotoxic effects by studying the difference in selectivity of the three cell lines to the compounds. As a result, pyrazole-benzimidazole hybrids may lower cell cycle regulators, including cyclin D1 and CDK2 in MCF-7 cells, causing cell cycle arrest and growth suppression [69] (refer to Figure 5 for Structure and SAR). Because the pyrazolo[3,4-d]pyrimidine nucleus is an isostere of the purine nucleus, it has an anticancer effect via serving as an ATP competitive inhibitor for several kinase enzymes. It has been reported that hydrazinyl derivatives have anticancer properties, particularly in breast cancer cell lines. There is evidence that a methyl sulphonyl ring at position 3 enhances the antitumor activity of pyrazolo[3,4-d]pyrimidine nucleic. Compound 6d (X = H, R = 4-F) showed the highest IC50 at 7.5 nM. More research is needed to understand the precise mechanism of antitumor activity and investigate the SAR of various nucleus locations [94] (refer to Figure 5 for Structure and SAR). A high-throughput screening method identified the pyrazolo[1,5-a]pyrimidine scaffold as a potent Jak2 inhibitor. Increased polarity and the elimination or inhibition of metabolic hotspots such as N-methyl groups are two often used approaches for increasing stability against CYP-mediated metabolism. The H-bond accepting properties of oxygen atoms in heteroaromatic ring systems are weak; on the other hand, sp2 nitrogens of heteroaromatics tend to be strong H-bond acceptors. Compound 84 was found to be the most promising derivative, with an IC50 of 7.4 nM. Compound 84 offers a good combination of cell potency, oral exposure, and in vivo pSTAT5 knockdown, making it a feasible tool for initial Jak2-dependent efficacy studies. Any alteration at the para-position of the N-aryl group reduces the inhibitory potency of all Jak family members considerably. The pyrazolo[1,5-a]pyrimidine scaffold was shown to be a strong inhibitor of Jak2 in a high-throughput screening study [98] (refer to Figure 5 for Structure and SAR).
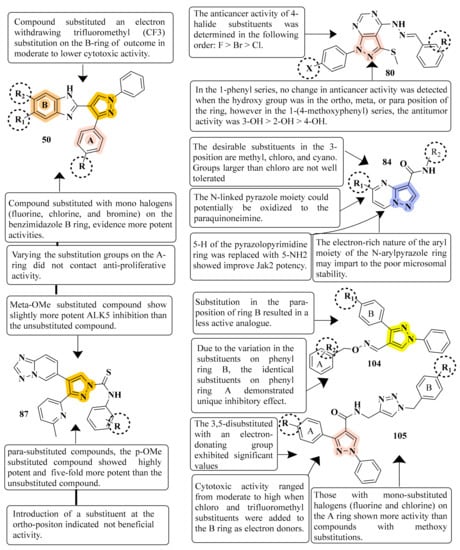
Figure 5. Structure and SAR of potent anticancer activity compounds 50, 80, 87, 104, and 105.
Several of the pyridyl-substituted five-membered heterocyclic rings were boosted in their inhibitory activity and selectivity by adding methylene, methylene amino, or amino methylene linkages between their core rings and their phenyl rings. The most active compound 87 inhibited ALK5 phosphorylation with a IC50 value of 0.57 nM and demonstrated 94 percent inhibition at 100 nM in a luciferase reporter test using HaCaT cells irreversibly transfected with a p3TP-Luc construct. There was a significant increase in both the inhibitory activity and selectivity of compound 87 against p38a MAP kinase upon the incorporation of the [1,2,4]triazolo[1,5-a]pyridin-6-yl and phenycarbothioamide moiety at positions 4 and 1 of the pyrazole ring, respectively [101] (refer to Figure 5 for Structure and SAR).This work demonstrated the strong immunosuppressive actions of a variety of new pyrazole derivatives having an O-benzyl oxime moiety. Compound 104 had the greatest inhibitory efficacy (IC50 = 1.18 µM for lymph node cells and IC50 = 0.28 µM for PI3K). In particular, a variety of synthesized pyrazole oxime ether analogs were shown to have significant immunosuppressive effects in the low micromolar range. A study of the A-ring para substituents revealed that an electron-withdrawing group had increased immunosuppressive action, and the potency order is F > Cl > OMe > H > Me. The molecules with a potent inhibition effect towards IL-6 produced in ConA-stimulated murine lymph node cells were investigated [117] (refer to Figure 5 for Structure and SAR). The synthesis of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1,3-diphenyl-1H-pyrazole-4-carboxamides was reported well as their biological evaluation. Nocodazole (8.01–0.95 µM) exhibited significant inhibition against MIAPaCa-2, MCF-7, and HeLa cell lines with compound 105 (GI50) in the range of 0.13–0.7 µM (GI50). Using the pyrazole moiety with two aryl ring systems, A and B, this study investigated the compounds with electron-withdrawing and electron-donating groups, i.e., chloro, fluoro, and fluoro trifluoromethyl groups on the ring systems (A and B). This compound 105 resulted in cell cycle arrest in MCF-7 cells and downregulation of CDK1 expression in MCF-7 cells when it was treated with it. As a result, they can be regarded as promising lead molecules for creating more effective anticancer medicines against breast cancer cells [118] (refer to Figure 5 for Structure and SAR).
APN inhibition was best with compound 109, which had an IC50 value of 0.16 ± 0.02 μM, a value over one order of magnitude less than that of bestatin (IC50 = 9.4 ± 0.5 μM). Depending on where R was substituted on the pyrazoline ring, these compounds could have significantly different APN inhibitory potencies. B series compounds have higher inhibitory activity when compared to APN equivalents. As determined by the structure–activity relationships (SARs) of A series compounds, only ortho- or meta-substituted hydroxamates can be synthesized from B series compounds, while para-substituted hydroxamates are inactive. Compound 109 might be a starting point for developing more powerful APN inhibitors. To test human APN against the ES-2 and PLC/PRF/5 cell surfaces, compounds with significant porcine kidney APN inhibitory properties were selected. APN is abundantly expressed on both cell lines’ membranes [122] (refer to Figure 6 for Structure and SAR). The strongest inhibitory effect was achieved by compound 111 with N-(3-methoxy-2-hydroxybenzal)-3-substituted(p-chlorophenyl)-4-cyano-5-oxopyrazol-1-thiocarboxamide against HL-60, with an IC50 of 1.35 μM compared to Dox’s IC50 of 2.02 μM. Compound 111 improved the G2/M phase from 11.05 percent to 39.22 percent compared to the untreated control. The highest intense cytotoxic action was demonstrated by compound 111. In a study that included activity assays of apoptosis detection and Topoisomerase II inhibition, compound 111 was found to be an effective inhibitor of Topo II [124] (refer to Figure 6 for Structure and SAR).
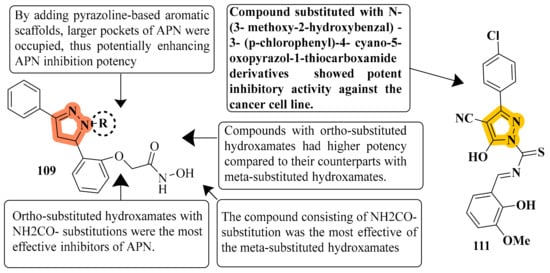
Figure 6. Structure and SAR of potent anticancer activity compounds 109 and 111.
3. Pyrazole Biomolecules as Inflammation Therapeutics
The efficacy of new pyrazole and pyrazoline compounds to inhibit ovine COX-1 and COX-2 isozymes was assessed utilizing an in vitro cyclooxygenase (COX) inhibition test. Among the molecules examined, 129 exhibited excellent COX-2 inhibitory efficacy (IC50 = 0.26 µM) and selectivity (SI) = >192.3, which is equivalent to the reference medication celecoxib (IC50 = 0.28 µM and selectivity index = 178.57). Compound 129 is overly lipophilic due to the presence of trifluoromethyl fragments; therefore, it should serve as a lead molecule for further optimization of certain biological properties and ADME characteristics [127] (refer to Figure 7 for Structure and SAR). Novel 1-(4-methane(amino)sulfonylphenyl) derivatives 5th (4-substituted-aminomethyl-phenyl)-3-trifluoromethyl-1H-pyrazoles were developed, synthesized, and tested for biological activity. Compound 168, derived from the library, exhibited significant anti-inflammatory action. Compared with the standard celecoxib drug (80% inhibition), compound 168 demonstrated the greatest anti-inflammatory effect post carrageenan, 3 h after inflammation onset (89% inhibition) [179]. A total of fifteen pyrazolyl urea derivatives were produced and tested for their ability to inhibit p38 MAPK and act as antioxidants. Among the tested compounds, derivative 178 was discovered to be the most powerful, and its binding mechanism inside the p38 MAPK was also documented. Compound 178, which has a 4- chloro group and has significant p38 MAPK activity, also had the greatest anti-inflammatory efficacy (80.93 percent inhibition). The substitution of 2-chloro and 3-chloro groups for the 4-chloro groups resulted in a modest reduction in inactivity (76.19 percent and 78.06 percent inhibition, respectively). In both the in vitro and in vivo models, the monosubstituted electron-withdrawing group in the phenyl ring linked to the pyrazole nucleus showed strong anti-inflammatory efficacy. Compound 178 was found to be a strong anti-inflammatory agent, equivalent to the standard reference medication diclofenac sodium. In addition, compared to conventional medicine, this molecule had a lower ulcerogenic potential and the least activity in causing oxidative stress in tissues. In contrast, pyrazole compounds with a urea pharmacophore exhibited comparable anti-inflammatory effects and better TNF-inhibitory characteristics in the investigations. Moreover, pyrazolyl urea derivatives were investigated for p38 MAPK inhibition and demonstrated substantial efficacy [187] (refer to Figure 7 for Structure and SAR).
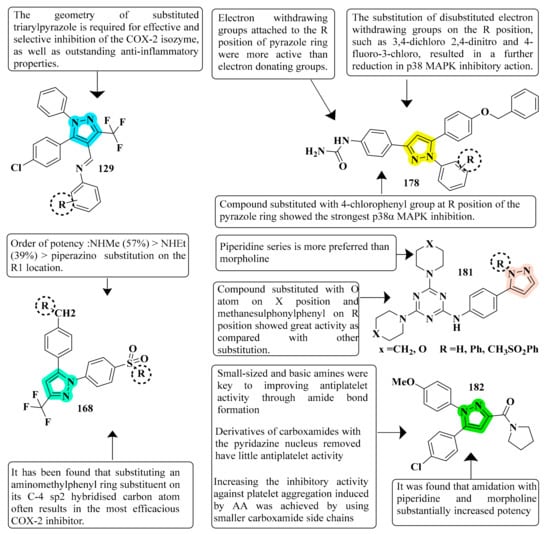
Figure 7. Structure and SAR of potent anti-inflammatory activity compounds 129, 168, 178, 181, and 182.
An inhibitor of COX-2 with a five-membered pharmacophore, such as pyrazole, and isoxazole, which is polysubstituted 1,3,5-triazines. Compound 181 from synthesized derivatives showed promising findings with an IC50 of 0.74 μM vs. celecoxib’s IC50 of 0.78 μM. When a methanesulphonylphenyl pyrazole moiety was added, the selectivity increased significantly. To summarize, when the triazine core was replaced with pyrazoline, isoxazole, or unsubstituted pyrazole moieties, a considerable COX-2 selectivity was observed. Thus, scaffolds derived from Compound 181 might be further developed, and their biological activity tested [190] (refer to Figure 7 for Structure and SAR). The pyridazine 6-position was substituted with phenyl, and the carboxyl arm in the central pyrazole was extended by adding polar substituents. In addition, pyrazole compounds without either the 1-pyridazine or the 5-phenyl groups were synthesized. These derivatives were designed to explore how a side chain of different sizes and basicity at three positions of the pyrazole could affect the antiplatelet activity and to learn about the effects of exposure to vicinal diaries about the central heterocycle, as well as the necessary presence of central pyrazoles on their biological profiles. Various compounds of this class had different inhibitory effects on platelet aggregation induced by AA and collagen, mainly depending on the shape and size of the carboxamide molecules at the 3-position of the pyrazole ring. However, only small carboxamide molecules specifically inhibited collagen-induced platelet aggregation. Finally, researchers describe compound 182 as a new potent antiplatelet agent with an IC50 in the two- to one-digit nanomolar range (IC50 values of 5.7 nM). Because of their great efficacy in the cellular milieu, these simple pyrazole derivatives may hold promise for developing more effective molecules for cardiovascular disease intervention [191] (refer to Figure 7 for Structure and SAR).
The primary pyrazole core of selective COX2 inhibitors (celecoxib and SC558) was engaged, and structural alterations led to the identification of additional pyrazole analogs. In comparison to ibuprofen (which inhibited COX-1/2 and TNF- after 4 h and exhibited 81.32 percent inhibition), compound 187 exhibited 80.63 percent inhibition at 4 h. We, therefore, found that methylamine pyrazole derivatives provided the requisite geometry to effectively and selectively inhibit the COX2 enzyme and provide good anti-inflammatory properties when substituted for sulfonamide pyrazole derivatives. As a result, the hydrophobic benzyloxyphenyl group was introduced in the same way as the celecoxib trifluoromethyl group to boost the affinity for COX2 [196] (refer to Figure 8 for Structure and SAR). A novel pyrazole derivative was created and evaluated for its potential to inhibit COX-1/2 activity. There was good selectivity towards COX-2 inhibition in many pyrazoles bearing the benzenesulfonamide moiety, and consequently, these compounds had powerful anti-inflammatory activity. Compounds 189(a), 189(b), 189(c), and 189(d) showed high activity and selectivity against COX-2 (IC50 = 39.43, 61.24, 38.73, and 39.14 nM). All compounds reported selectivity indices of 22.21, 14.35, 17.47, and 13.10 for 189(a), 189(b), 189(c), and 189(d), correspondingly. In vivo, these compounds were superior to or equivalent to celecoxib as anti-inflammatory drugs. Interestingly, these molecules specifically reduced the induced microsomal PGE2 synthase synthesis, indicating that additional research into these derivatives is needed [198] (refer to Figure 8 for Structure and SAR).
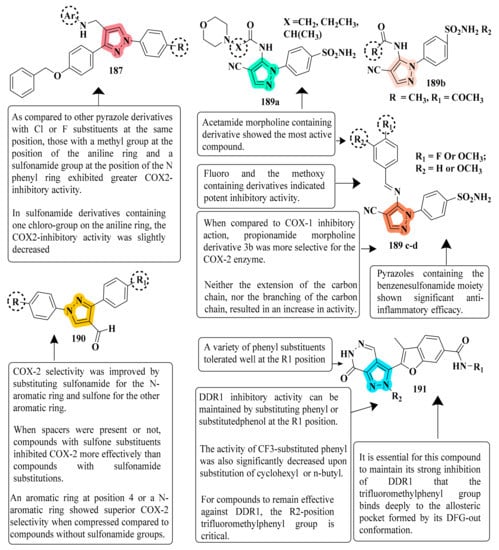
Figure 8. Structure and SAR of potent anti-inflammatory activity compounds 187, 189, 190 and 191.
In an easy synthetic procedure, a series of diaryl pyrazole and triazole derivatives to produce selective COX-2 inhibitors were designed and synthesized. Among the synthesized derivatives, compound 190a with a sulfonamide substitution on one N-aromatic ring had the strongest inhibitory action against COX-2 with IC50 = 0.017 ± 0.001 μM and COX-1 with IC50 = 0.263 ± 0.016 μM. In contrast, compound 190b, which has nitro on its N-aromatic ring, inhibited COX-1 more effectively (IC50 = 0.012 ± 0.001 μM). The best COX-2 selectivity was obtained with sulfonamide and sulfone substituted on the N-aromatic ring. More interestingly, the heterocyclic cores (pyrazole or triazole) contain two not vicinal aryls in contrast with the most commonly known COX-2 inhibitors. Additional work is needed to synthesize diverse pyrazoles and triazoles with different spacer lengths and substitutions in order to assess their role in COX-2 inhibitory activity and selectivity [199] (refer to Figure 8 for Structure and SAR). Researchers developed and tested new compounds known as pyrazolo[3,4-d]pyridazinones as anti-inflammatory drugs against discoidin domain receptor 1 (DDR1) modifications. Mouse bone-marrow-derived and human THP-11 macrophages were effectively decreased by compound 191 at ten micromolar concentrations when exposed to LPS. Compound 191 inhibited pro-inflammatory cytokines and autophosphorylation of DDR1 in cells in a mouse colitis model induced by dextran sulfate sodium (DSS). DDR1-IN-1, a positive control compound, had an EC50 of 114.5 nM, whereas compound 191 suppressed basal autophosphorylation at 34.4 nM, more powerful than the positive control compound. As the N-phenyl group of DC-1 was exposed above the solvent-exposed area of DDR1, further modifications can be made to this group in DDR1 [200] (refer to Figure 8 for Structure and SAR).
This entry is adapted from the peer-reviewed paper 10.3390/molecules27248708
This entry is offline, you can click here to edit this entry!