Colorectal cancer (CRC) represents one of the most common causes of death among cancers worldwide. Its incidence has been increasing among the young population. Many risk factors contribute to the development and progression of CRC and about 70% of them are sporadic. The CRC microenvironment is highly heterogeneous and represents a very complex immunosuppressive platform. Many cytokines and their receptors are vital participants in this immunosuppressive microenvironment. Tumor necrosis factors (TNFs) and TNF receptor 2 (TNFR2) are critical players in the development of CRC. TNFR2 was observed to have increased the immunosuppressive activity of CRC cells via regulatory T cells (T regs) and myeloid-derived suppressor cells (MDSC) in the CRC microenvironment.
1. Introduction: Overview of Colorectal Cancer
Colorectal cancer (CRC) is the third most common cancer in men after lung and prostate, with about 1.06 million new cases reported in 2020. It is the second most common cancer in women after breast cancer, with a reported 865,630 new cases
[1]. In the past, CRC has primarily affected the elderly and was often diagnosed in the late stages of the disease; its prevalence among young people is currently growing
[2]. CRC is a heterogeneous disease and about 10% of all adenomas develop into invasive cancer; 95% of all CRC that results from these adenomatous polyps is referred to as an adenocarcinoma
[3]. CRC develops over time, approximately 10 to 15 years, due to a series of well-defined signaling pathways and genetic and epigenetic changes that lead to the inactivation and activation of tumor suppressor genes and oncogenes, respectively. Additionally, CRC is caused by three key mechanisms: chromosomal instability (CIN), microsatellite instability (MSI), and the CpG island methylator phenotype (CIMP). Cancer genome aberrations and the formation of an inflammatory microenvironment are critical in the development and progression of CRC
[4][5]. Moreover, a favorable family history is seen in 10–20% of CRC patients. A detailed family history is critical in attaining a diagnosis, since it provides the most effective monitoring tool for CRC prevention. Non-polyposis (Lynch) and polyposis (Familial adenomatous polyposis) syndromes are the two types of hereditary CRC. Furthermore, modifiable variables and environmental lifestyles such as smoking, obesity, sedentary lifestyle, alcohol use, and a high diet of processed meat may raise the risk of CRC. The signs and symptoms vary among patients, but the most common symptoms are rectal bleeding, changes in bowel habit, anemia, or abdominal pain. The current screening options such as fecal occult blood test (FOBT), endoscopy, colonoscopy and CT colonoscopy are available for patients, depending on the patient’s needs and condition. Surgery is still the “gold standard” for CRC treatment and post-operative chemotherapy, immunotherapy, and radiotherapy may help to further improve patient survival and reduce the risk of cancer recurrence
[6].
In a normal microenvironment, to maintain immunological homeostasis, the immune system is activated during pathogen invasion, which results in the removal of such invaders from the host as well as the prevention of tumor formation via apoptosis. This initial reaction to pathogens relies on innate immunity, which is essential in protecting the host before adaptive immune responses arise. Unlike innate immunity, adaptive immunity develops in response to infection and detects a wide range of microbial and non-microbial agents
[7]. Humoral immunity and cell-mediated immunity are two types of adaptive immunity that are mediated by different types of lymphocytes and eradicate various microbes. Cell-mediated immunity is mediated by T lymphocytes and their products such as cytokines, CD4
+ T lymphocytes that assist macrophage to consume microorganisms and C8
+ CTL to make antibodies. But certain T lymphocytes such as regulatory T cells (Treg) have a function in suppressing immune response
[7]. Meanwhile, antibodies generated by B lymphocytes, plasma cells, and defense mechanisms against external microorganisms drive the humoral response
[7]. Immune cells also detect tumor-specific antigens or chemicals that are produced as a result of cellular stress.
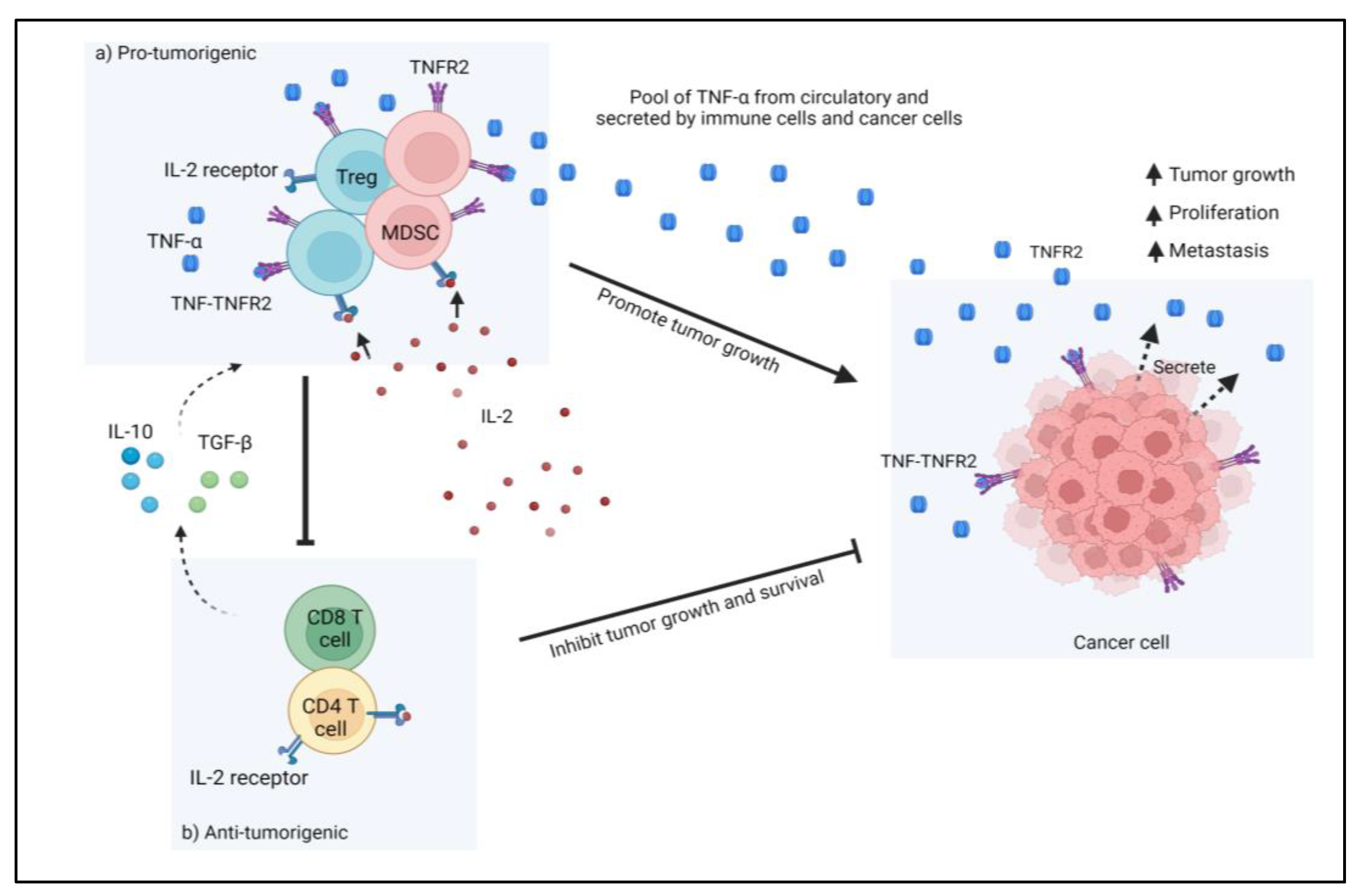
In contrast, this stress is detrimental in the tumor microenvironment (TME), where it leads to immune escape mechanisms of tumor cells and promotes tumor development. Additionally, when immune cells such as regulatory T cells (Treg) and myeloid-derived suppressive cells (MDSC) are enriched within the TME, they will dampen the activity of anti-tumor effectors and encourage tumor development. Moreover, the presence of Tregs and MDSCs has a significant suppressive role by inhibiting effector T cell activities and reducing CD4
+ T cells and effector CD8
+ T cells anti-tumor activation. Apart from that, other factors such as cell-surface proteins, cytokine/chemokine, transcriptional factors, and enzymes may contribute to the immunosuppressive role in the TME of CRC. These factors have the potential to be used as immunotherapy targets, such as PD-1/PD-L1, CTLA-4, which is expressed on Treg. They produced promising outcomes in CRC. Besides, interleukin-2 (IL-2) is important in CRC TME, where it is essential for the activation and proliferation of conventional T cells (Tconv) as well as the maintenance of Treg function and phenotype. However, Tregs do not make IL-2 on their own as they require external IL-2 for cell survival by increasing the affinity of the IL-2 receptor (CD25) to capture IL-2 from the environment, and so the amount of IL-2 for conventional T cells will be reduced
[8]. Cancer cells boost their potential for IL-2 absorption to evade the immune system. The presence of the cytokines IL-10 and TGF-β also promotes the activation, proliferation, and suppressive actions of Tregs and MDSCs, thereby enhancing their IL-2 uptake and creating conditions that are more conducive to tumor development and metastasis.
Figure 1 shows the action of immunosuppressive cells in TME that led to cancer progression. Furthermore, CRC tumor cells also produce TNFR2 on their surface, which interacts with TNF-α and promotes tumor cell proliferation in TME. However, studies are still ongoing, as TME immunity is complicated. The complexities of TME immunity encouraged us to look into the role of TNFR2 in CRC immune resistance via its expression in various cell types and signaling pathways that promote CRC development.
Figure 1. In tumor microenvironments (TME), immunosuppressive cells such as Treg cells and MDSCs suppress the function of anti-tumorigenic cells such as T cells and effector T cells. Treg cells compete with T cells for IL-2, which is necessary for T cell activation. Additionally, the presence of the cytokines IL-10 and TGF-β also increases the activation, proliferation, and suppressive activities of Tregs and MDSCs, hence increasing their IL-2 uptake. Furthermore, the presence of TNFR2 on Tregs, MDSCs, and tumor cells creates a favorable environment for tumor survival and growth via an immune escape mechanism.
2. Tumor Necrosis Factor Receptor 2 (TNFR2): An Outlook in CRC
Upregulated production of TNFR2 on cancer cells could severely enhance the development of multiple myeloma, renal cell carcinoma, Hodgkin’s lymphoma, cutaneous non-Hodgkin’s lymphoma, ovarian cancer, and colon cancer
[9]. For example, Zhao et al. have presented the effect of TNFR2 on proliferation, in such a way that the receptor successfully regulated the Ki-67 expression
[10]. The growth of SW116 was highly proliferative in the presence of TNFR2, but the growth of HT29 was reduced when TNFR2 was silenced
[10]. Furthermore, TNFR2 affects signaling pathways that are also important for the development and progression of tumors, such as P13K/AKT and MPAK/ERK. According to
[10], TNFR2 participated in AKT signaling by increasing the activated phosphorylation of AKT, not ERK
[10]. This indicates that TNFR2 can regulate the proliferation of CRC cells and promote the progression of CRC growth via P13K/AKT signaling pathway
[10].
In addition, Hamilton et al. showed that TNFR2 overexpression increased the proliferation of two different colon cancer cell lines. Overexpression of TNFR2 in COLO205 cells promoted anchorage-independent growth. Although the effects of TNFR2 overexpression on proliferation and anchorage-independent growth seem to be minor, it is crucial to note that the colon cancer cell lines employed have high basal proliferation rates and are often resistant to growth increase in response to external stimuli
[11]. The study further verified the induction of TNFR2 mRNA and protein by combined IL-6 and TNF, which offered direct evidence for the major function of STAT3 in TNFR2 induction. It also showed that TNF promoted IL-6 in SW480 cells, implying that TNF-induced IL-6 performs autocrine actions that contribute to the capacity of TNF and IL-6 to jointly drive TNFR2 expression
[11]. The study established that TNFR2 was raised during the episodes of acute dextran sodium sulphate (DSS)-colitis, which was preceded by IL-6/STAT3 activation.
Mizoguchi et al. presented the first evidence for the upregulation of the TNFR2 during intestinal inflammation
[12]. They also found that disrupting TNFR2 reduced the proliferation of intestinal epithelial cells (IEC) in a T-cell receptor (TCR) deficient colitis model
[12]. Previous in vitro investigations had shown that both IL-6 and TNF were necessary to trigger TNFR2 in CRC cells, perhaps indicating a physiological microenvironment of several cytokines in inflammatory bowel disease (IBD) or IBD-associated CRC. As such, the involvement of the immunosuppressive immune cells (such as Treg cells and MDSCs) also contributes to poor CRC prognosis. The higher levels of FOXP3 Treg cells are associated with poor prognosis, indicating that FOXP3 Treg cells inhibit tumor immune surveillance and so promote tumor development and progression
[13]. Furthermore, the presence of TNFR2 was seen on immunosuppressive cells such as Tregs, MDSCs, and also in the cancer cells. The expression of TNFR2 on these cells enhanced the suppressive functions and inhibited the antitumor action of effector cells, leading to tumor progression.
3. Targeting TNFR2 in CRC Cancer
TNFR2 acts as both an oncogene and Treg- or MDSCs-cells inducer. Furthermore, its limited expression on other cell types renders this receptor a selective target for cancer treatment. In respect to CRC, TNFR2 is responsible for the promotion of proliferation and angiogenesis through several other mechanisms, such as by mediating progranulin
[14] and STAT3
[11]. Currently, in a study with murine colon cancer models, the combination of anti-programmed death receptor-1 with new anti-TNFR2 led to complete tumor regression and elimination in more than 60% of the animals
[15]. The mechanism behind this therapeutic potency is thought to involve the reduction of immunosuppressive Treg cells in the TME, thus increasing the ratio of the CD8
+ effector cells to the Treg cells. The role of TNFR2 in CRC, however, is not only associated with its functional and preferential expression to Treg cells. Soluble TNFR2 (sTNFR2) reflects the upregulation of TNFR2 during inflammation, and the elevation of sTNFR2 was shown to increase the risk of CRC
[16]. Additionally, higher sTNFR2 levels in patients with CRC are associated with higher mortality, although not CRC-specific mortality
[17]. These roles of TNFR2 in the pathogenesis and progression of CRC render this receptor a potent target for CRC therapy. For cancers, including CRC, inhibition of TNFR2 or depletion of TNFR2-expressing cells can be achieved with the conventional antibody technology, and both in vitro and in vitro observations have been encouraging
[15][18][19][20]. Although this approach could produce high-affinity anti-TNFR2, several limitations are of concern, including the unwanted FcγR-receptor side effects and the fact that it is only effective in the dominant manner of TNFR2 in the expansion of Treg cells
[21]. TNFR2 is established to be preferentially expressed on Treg cells and important for their suppressive function; however, these characteristics are not exclusive to TNFR2 alone. Several other receptors, such as CD28 and CD25, as well as other TNFR receptors (GITR, OX40, 4-1BB), could also promote either the proliferation or the function of Treg cells, thus limiting the efficacy of targeting against any of these molecules. A better approach would be a system of targeting Treg cells as a whole by integrating all or most of their receptors. One such system is conceivably possible with the advent of nanotechnologies. Nanotechnologies have grown over the decades into a highly prospective tool in cancer therapy, where they are routinely used as drug and immune modulators and carriers that provide targeted delivery and prolonged accumulation in the organism
[22].
Therefore, nanotechnologies may provide enhanced response rates and reduce the side effects that limit the full capacities of conventional drugs. Nanotechnology-based therapy can overcome the disadvantages of conventional therapy because of its small size, non-toxicity, biocompatibility, and distinct physicochemical properties
[6]. Nanomaterials can be made comparable in size to biological macromolecules, making them ideal for cancer detection and treatment
[6]. In addition, the advancement of nanotechnology-based medicinal approaches has resulted in significant improvements in the solubility of drugs as well as in their efficacy, bioavailability, and pharmacokinetics. As such, several nanomedicines have been approved by the FDA, such as liposomes
[6] for cancer management, especially in the metastasis stage, and have been shown to fulfill their advantageous prospects, although there are still limited options existing for CRC
[23].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11010173