Nitrogen–fixing bacteria execute biological nitrogen fixation through nitrogenase, converting inert dinitrogen (N2) in the atmosphere into bioavailable nitrogen. Elaborating the molecular mechanisms of orderly and efficient biological nitrogen fixation and applying them to agricultural production can alleviate the “nitrogen problem”. Azotobacter vinelandii is a well–established model bacterium for studying nitrogen fixation, utilizing nitrogenase encoded by the nif gene cluster to fix nitrogen. In Azotobacter vinelandii, the NifA–NifL system fine–tunes the nif gene cluster transcription by sensing the redox signals and energy status, then modulating nitrogen fixation.
- biological nitrogen fixation
- nitrogenase
- NifA–NifL system
- biological nitrogen fertilizer
1. Introduction
2. Nitrogenase and Its Transcriptional Regulation
2.1. Nitrogenase
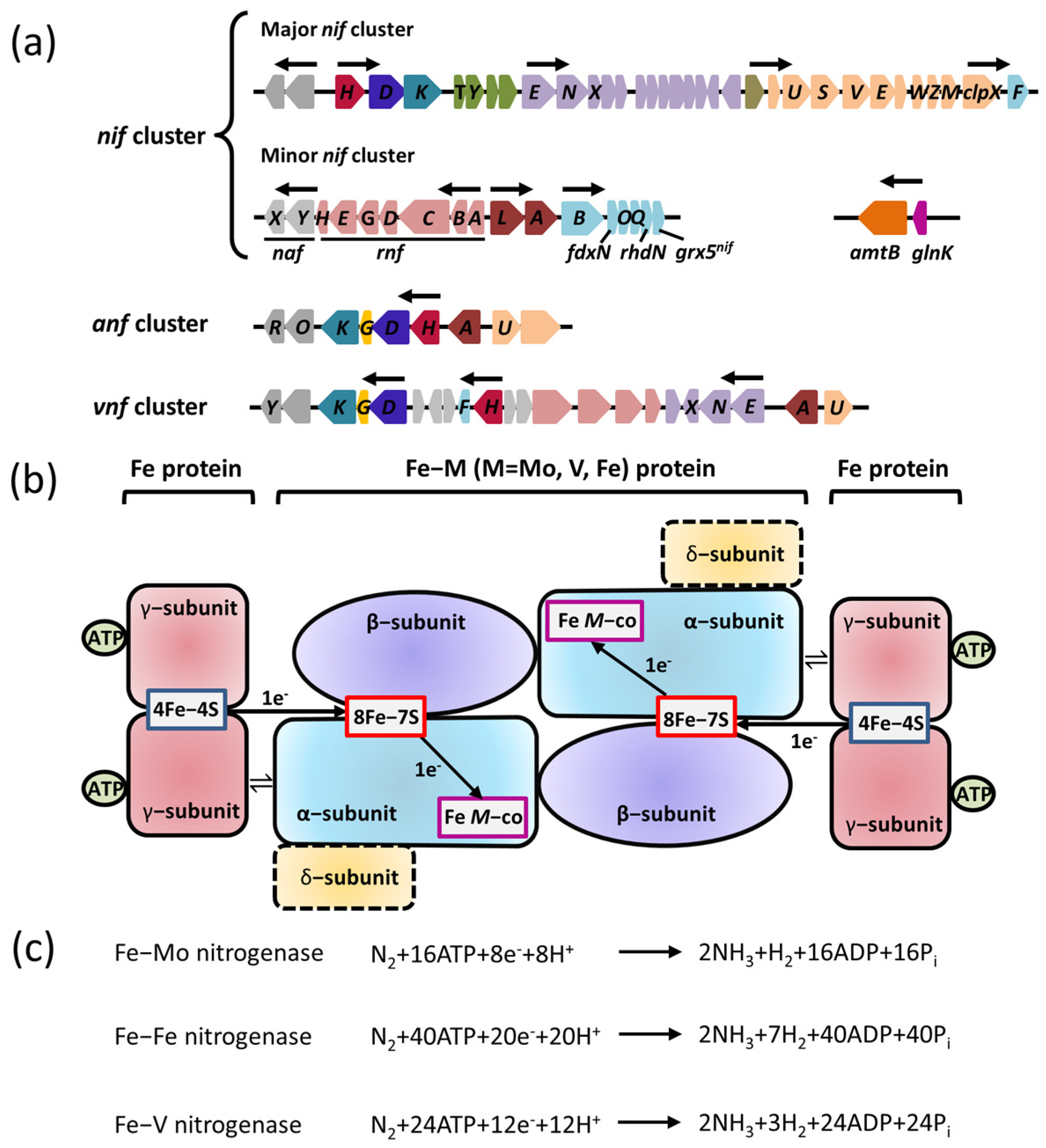
2.2. Transcriptional Regulation of Nitrogenase

3. The NifL–NifA System Responds to the Transcriptional Regulation of Nitrogenase via Environmental Signaling Molecules

3.1. Regulation of NifA Function by 2–OG
3.2. Effects of ADP and FAD Molecules on NifL Function
3.3. The NifL–NifA System Regulated by GlnK
This entry is adapted from the peer-reviewed paper 10.3390/ijms24020907
References
- Stokstad, E. The nitrogen fix. Science 2016, 353, 1225–1227.
- Fan, K.; Delgado-Baquerizo, M.; Guo, X.; Wang, D.; Wu, Y.; Zhu, M.; Yu, W.; Yao, H.; Zhu, Y.G.; Chu, H. Suppressed N fixation and diazotrophs after four decades of fertilization. Microbiome 2019, 7, 143.
- Bonilla Cedrez, C.; Chamberlin, J.; Guo, Z.; Hijmans, R.J. Spatial variation in fertilizer prices in Sub-Saharan Africa. PLoS ONE 2020, 15, e0227764.
- Ye, Y.; Ngo, H.H.; Guo, W.; Chang, S.W.; Nguyen, D.D.; Varjani, S.; Ding, A.; Bui, X.T.; Nguyen, D.P. Bio-membrane based integrated systems for nitrogen recovery in wastewater treatment: Current applications and future perspectives. Chemosphere 2021, 265, 129076.
- Kumar, M.; Tomar, R.S.; Lade, H.; Paul, D. Methylotrophic bacteria in sustainable agriculture. World J. Microbiol. Biotechnol. 2016, 32, 120.
- Canfield, D.E.; Glazer, A.N.; Falkowski, P.G. The evolution and future of Earth’s nitrogen cycle. Science 2010, 330, 192–196.
- Hu, Y.; Ribbe, M.W. Biosynthesis of the Metalloclusters of Nitrogenases. Annu. Rev. Biochem. 2016, 85, 455–483.
- Das, H.K. Azotobacters as biofertilizer. Adv. Appl. Microbiol. 2019, 108, 1–43.
- Romero-Perdomo, F.; Abril, J.; Camelo, M.; Moreno-Galvan, A.; Pastrana, I.; Rojas-Tapias, D.; Bonilla, R. Azotobacter chroococcum as a potentially useful bacterial biofertilizer for cotton (Gossypium hirsutum): Effect in reducing N fertilization. Rev. Argent. Microbiol. 2017, 49, 377–383.
- Zambrano-Mendoza, J.L.; Sangoquiza-Caiza, C.A.; Campaa-Cruz, D.F.; Yánez-Guzmán, C.F. Use of Biofertilizers in Agricultural Production. In Technology in Agriculture; IntechOpen: London, UK, 2021; ISBN 978-1-83881-921-7.
- Hakeem, K.R.; Akhtar, J.; Sabir, M.J.S.I.P. Azotobacter chroococcum—A Potential Biofertilizer in Agriculture: An Overview. In Soil Science: Agricultural and Environmental Prospectives; Springer: Cham, Switzerland, 2016; pp. 333–348.
- Bueno Batista, M.; Dixon, R. Manipulating nitrogen regulation in diazotrophic bacteria for agronomic benefit. Biochem. Soc. Trans. 2019, 47, 603–614.
- Wilson, P.W.; Burris, R.H.; Lind, C.J. The Dissociation Constant in Nitrogen Fixation by Azotobacter. Proc. Natl. Acad. Sci. USA 1942, 28, 243–250.
- Seefeldt, L.C.; Hoffman, B.M.; Dean, D.R. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 2009, 78, 701–722.
- Harwood, C.S. Iron-Only and Vanadium Nitrogenases: Fail-Safe Enzymes or Something More? Annu. Rev. Microbiol. 2020, 74, 247–266.
- Rutledge, H.L.; Cook, B.D.; Nguyen, H.P.M.; Herzik, M.A., Jr.; Tezcan, F.A. Structures of the nitrogenase complex prepared under catalytic turnover conditions. Science 2022, 377, 865–869.
- Seefeldt, L.C.; Yang, Z.Y.; Duval, S.; Dean, D.R. Nitrogenase reduction of carbon-containing compounds. Biochim. Biophys. Acta 2013, 1827, 1102–1111.
- Rubio, L.M.; Ludden, P.W. Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 2008, 62, 93–111.
- Boyd, E.S.; Hamilton, T.L.; Peters, J.W. An alternative path for the evolution of biological nitrogen fixation. Front. Microbiol. 2011, 2, 205.
- Wang, L.; Zhang, L.; Liu, Z.; Zhao, D.; Liu, X.; Zhang, B.; Xie, J.; Hong, Y.; Li, P.; Chen, S.; et al. A minimal nitrogen fixation gene cluster from Paenibacillus sp. WLY78 enables expression of active nitrogenase in Escherichia coli. PLoS Genet. 2013, 9, e1003865.
- Curatti, L.; Brown, C.S.; Ludden, P.W.; Rubio, L.M. Genes required for rapid expression of nitrogenase activity in Azotobacter vinelandii. Proc. Natl. Acad. Sci. USA 2005, 102, 6291–6296.
- Arnold, W.; Rump, A.; Klipp, W.; Priefer, U.B.; Puhler, A. Nucleotide sequence of a 24,206-base-pair DNA fragment carrying the entire nitrogen fixation gene cluster of Klebsiella pneumoniae. J. Mol. Biol. 1988, 203, 715–738.
- Little, R.; Martinez-Argudo, I.; Dixon, R. Role of the central region of NifL in conformational switches that regulate nitrogen fixation. Biochem. Soc. Trans. 2006, 34, 162–164.
- Huergo, L.F.; Dixon, R. The Emergence of 2-Oxoglutarate as a Master Regulator Metabolite. Microbiol. Mol. Biol. Rev. 2015, 79, 419–435.
- Senior, P.J. Regulation of nitrogen metabolism in Escherichia coli and Klebsiella aerogenes: Studies with the continuous-culture technique. J. Bacteriol. 1975, 123, 407–418.
- Martinez-Argudo, I.; Little, R.; Dixon, R. Role of the amino-terminal GAF domain of the NifA activator in controlling the response to the antiactivator protein NifL. Mol. Microbiol. 2004, 52, 1731–1744.
- Little, R.; Dixon, R. The amino-terminal GAF domain of Azotobacter vinelandii NifA binds 2-oxoglutarate to resist inhibition by NifL under nitrogen-limiting conditions. J. Biol. Chem. 2003, 278, 28711–28718.
- Little, R.; Colombo, V.; Leech, A.; Dixon, R. Direct interaction of the NifL regulatory protein with the GlnK signal transducer enables the Azotobacter vinelandii NifL-NifA regulatory system to respond to conditions replete for nitrogen. J. Biol. Chem. 2002, 277, 15472–15481.
- Henry, J.T.; Crosson, S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 2011, 65, 261–286.
- Vogt, J.H.; Schippers, J.H. Setting the PAS, the role of circadian PAS domain proteins during environmental adaptation in plants. Front. Plant Sci. 2015, 6, 513.
- Hill, S.; Austin, S.; Eydmann, T.; Jones, T.; Dixon, R. Azotobacter vinelandii NIFL is a flavoprotein that modulates transcriptional activation of nitrogen-fixation genes via a redox-sensitive switch. Proc. Natl. Acad. Sci. USA 1996, 93, 2143–2148.
- Hill, S.; Kennedy, C.; Kavanagh, E.; Goldberg, R.B.; Hanau, R. Nitrogen fixation gene (nifL) involved in oxygen regulation of nitrogenase synthesis in K. pneumoniae. Nature 1981, 290, 424–426.
- Key, J.; Hefti, M.; Purcell, E.B.; Moffat, K. Structure of the redox sensor domain of Azotobacter vinelandii NifL at atomic resolution: Signaling, dimerization, and mechanism. Biochemistry 2007, 46, 3614–3623.
- Little, R.; Salinas, P.; Slavny, P.; Clarke, T.A.; Dixon, R. Substitutions in the redox-sensing PAS domain of the NifL regulatory protein define an inter-subunit pathway for redox signal transmission. Mol. Microbiol. 2011, 82, 222–235.
- Little, R.; Reyes-Ramirez, F.; Zhang, Y.; van Heeswijk, W.C.; Dixon, R. Signal transduction to the Azotobacter vinelandii NIFL-NIFA regulatory system is influenced directly by interaction with 2-oxoglutarate and the PII regulatory protein. EMBO J. 2000, 19, 6041–6050.
- He, S.; Chen, M.; Xie, Z.; Yan, Y.; Li, H.; Fan, Y.; Ping, S.; Lin, M.; Elmerich, C. Involvement of GlnK, a PII protein, in control of nitrogen fixation and ammonia assimilation in Pseudomonas stutzeri A1501. Arch Microbiol. 2008, 190, 1–10.
- Reyes-Ramirez, F.; Little, R.; Dixon, R. Role of Escherichia coli nitrogen regulatory genes in the nitrogen response of the Azotobacter vinelandii NifL-NifA complex. J. Bacteriol. 2001, 183, 3076–3082.
- Rudnick, P.; Kunz, C.; Gunatilaka, M.K.; Hines, E.R.; Kennedy, C. Role of GlnK in NifL-mediated regulation of NifA activity in Azotobacter vinelandii. J. Bacteriol. 2002, 184, 812–820.
- Oliveira, M.A.; Aquino, B.; Bonatto, A.C.; Huergo, L.F.; Chubatsu, L.S.; Pedrosa, F.O.; Souza, E.M.; Dixon, R.; Monteiro, R.A. Interaction of GlnK with the GAF domain of Herbaspirillum seropedicae NifA mediates NH(4)(+)-regulation. Biochimie 2012, 94, 1041–1047.
- Meletzus, D.; Rudnick, P.; Doetsch, N.; Green, A.; Kennedy, C. Characterization of the glnK-amtB operon of Azotobacter vinelandii. J. Bacteriol. 1998, 180, 3260–3264.
- Oliveira, M.A.; Gerhardt, E.C.; Huergo, L.F.; Souza, E.M.; Pedrosa, F.O.; Chubatsu, L.S. 2-Oxoglutarate levels control adenosine nucleotide binding by Herbaspirillum seropedicae PII proteins. FEBS J. 2015, 282, 4797–4809.
- Perry, S.; Shearer, N.; Little, R.; Dixon, R. Mutational analysis of the nucleotide-binding domain of the anti-activator NifL. J. Mol. Biol. 2005, 346, 935–949.
- Xu, Y.; Cheah, E.; Carr, P.D.; van Heeswijk, W.C.; Westerhoff, H.V.; Vasudevan, S.G.; Ollis, D.L. GlnK, a PII-homologue: Structure reveals ATP binding site and indicates how the T-loops may be involved in molecular recognition. J. Mol. Biol. 1998, 282, 149–165.
- Hesketh, A.; Fink, D.; Gust, B.; Rexer, H.U.; Scheel, B.; Chater, K.; Wohlleben, W.; Engels, A. The GlnD and GlnK homologues of Streptomyces coelicolor A3(2) are functionally dissimilar to their nitrogen regulatory system counterparts from enteric bacteria. Mol. Microbiol. 2002, 46, 319–330.
- Jiang, P.; Ninfa, A.J. Alpha-ketoglutarate controls the ability of the Escherichia coli PII signal transduction protein to regulate the activities of NRII (NrB but does not control the binding of PII to NRII. Biochemistry 2009, 48, 11514–11521.
- Jiang, P.; Ninfa, A.J. Sensation and signaling of alpha-ketoglutarate and adenylylate energy charge by the Escherichia coli PII signal transduction protein require cooperation of the three ligand-binding sites within the PII trimer. Biochemistry 2009, 48, 11522–11531.
- Martinez-Argudo, I.; Little, R.; Shearer, N.; Johnson, P.; Dixon, R. The NifL-NifA System: A multidomain transcriptional regulatory complex that integrates environmental signals. J. Bacteriol. 2004, 186, 601–610.