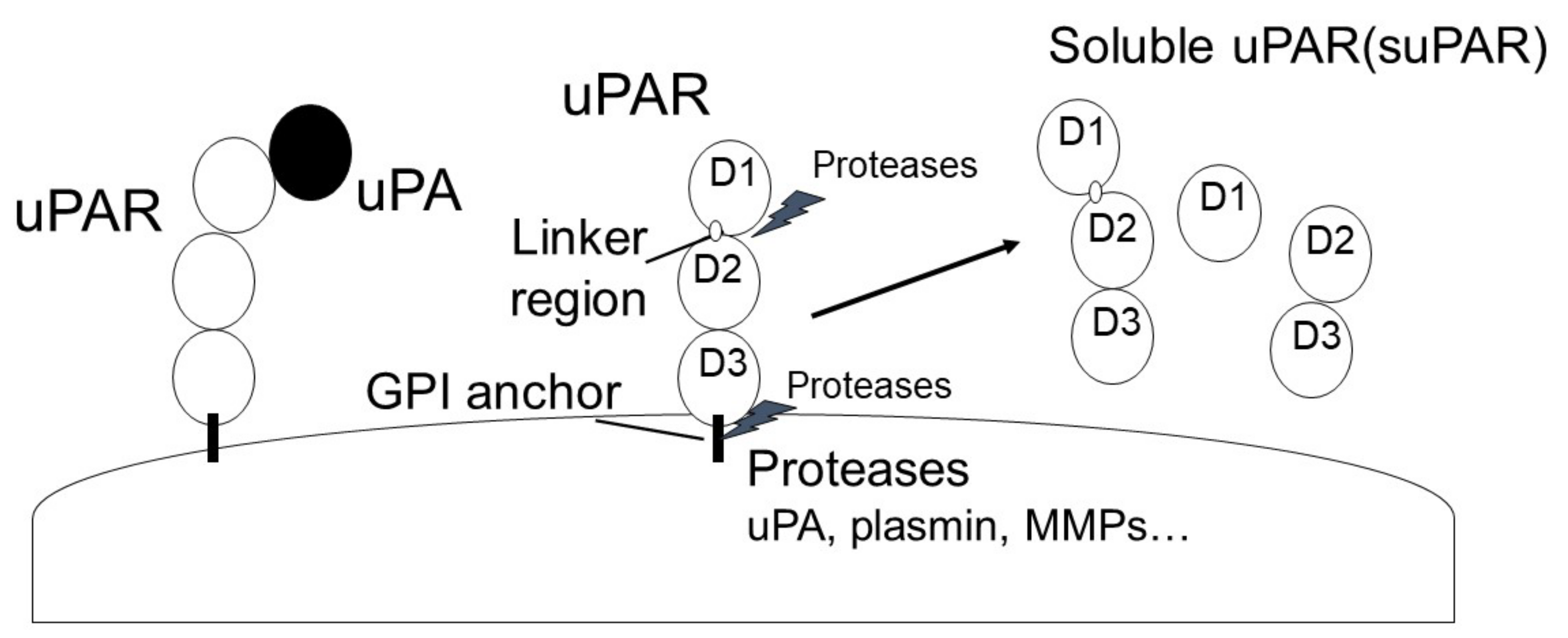
Urokinase plasminogen activator (uPA) is a single-chain serine protease that can cleave and activate Plg into plasmin by binding to urokinase plasminogen activator receptor (uPAR). uPA is secreted as a single-chain glycosylated zymogen called pro-uPA, and pro-uPA is activated by several proteinases, such as kallikrein, stromelysin, and plasmin. uPAR is a glycosyl-phosphatidyl-inositol anchored (GPI) membrane protein that consists of three domains: D1 (residues 1–92), D2 (residues 93–191) and D3 (residues 192–283). uPAR is cleaved between the D1 and D2 domains (linker region) and the GPI-anchor domain by several proteases, such as uPA, plasmin, MMPs, and GPI-specific phospholipase D, and then forms soluble uPAR (suPAR; full length D1-D3, D2D3, and D1).
- uPA
- uPAR
- plasmin
- fibrosis 23
1. The Urokinase Plasminogen Activator (uPA) and uPAR System
1.1. uPA
1.2. uPAR
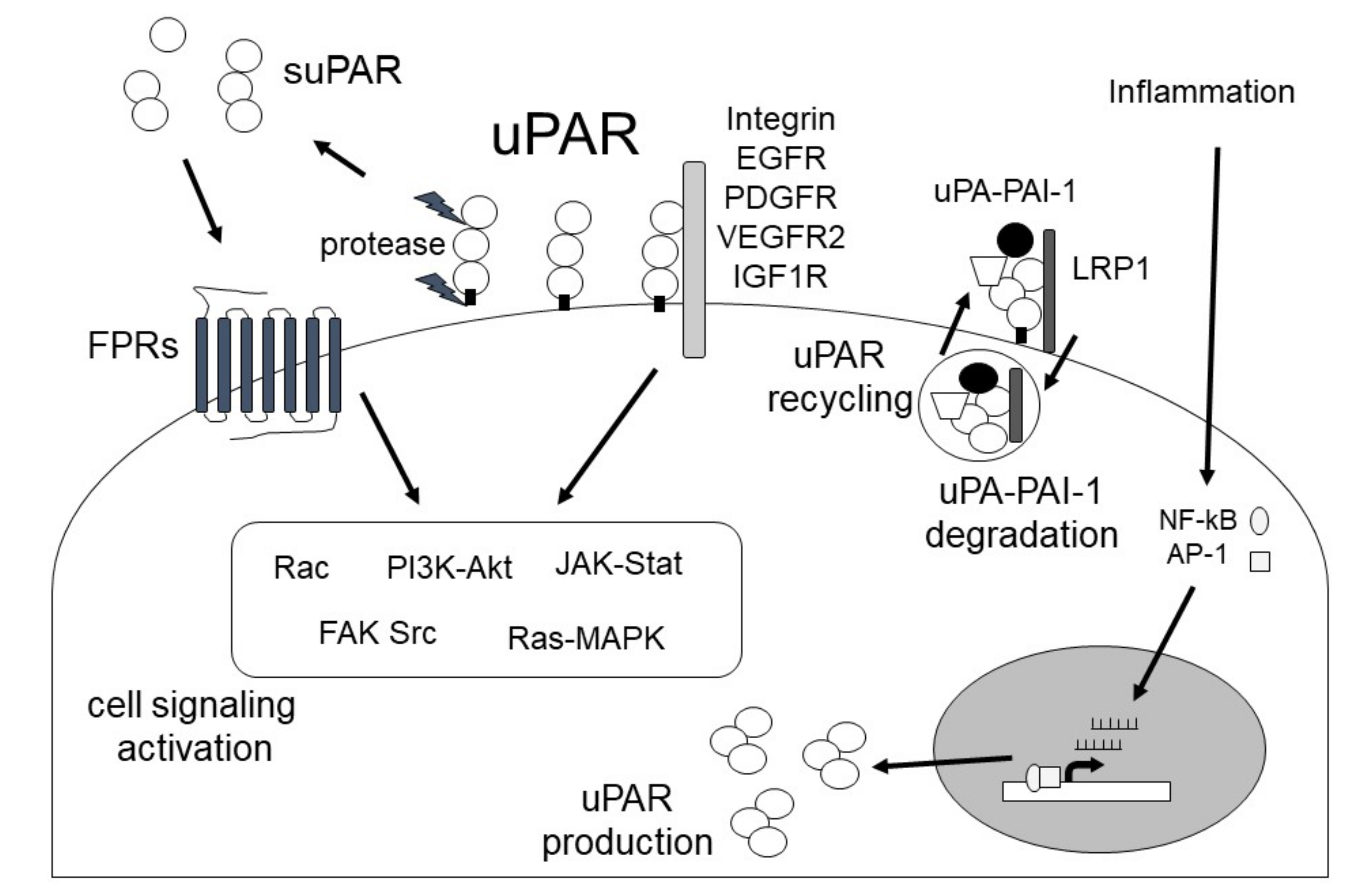
2. The Role of the uPA/uPAR System in Fibrosis
2.1. Myofibroblasts in Fibrosis
2.1.1. The uPA/uPAR System and Myofibroblasts
2.1.2. uPAR-Binding Protein and Myofibroblasts
2.1.3. Other Fibrinolytic Factors and Myofibroblasts
2.2. Suppression of ECM Depredating Protease in Fibrosis
3. Vascular Endothelial Dysfunction in Fibrosis
3.1. The Role of the uPA/uPAR System in EC Functions
3.2. The Role of the uPA/uPAR System in Angiogenesis
3.3. The Role of the uPA/uPAR System in Coagulation
3.4. The Role of the uPA/uPAR System in Vascular Tone Alteration and Hypertension
4. Immune Abnormalities and Inflammation in Fibrosis
5. The Role of the uPA/uPAR System in Inflammation and the Immune System
This entry is adapted from the peer-reviewed paper 10.3390/ijms24021796
References
- Kanno, Y.; Kaneiwa, A.; Minamida, M.; Kanno, M.; Tomogane, K.; Takeuchi, K.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The Absence of uPAR Is Associated with the Progression of Dermal Fibrosis. J. Investig. Dermatol. 2008, 128, 2792–2797.
- Kanno, Y.; Matsuno, H.; Kawashita, E.; Okada, K.; Suga, H.; Ueshima, S.; Matsuo, O. Urokinase-type plasminogen activator receptor is associated with the development of adipose tissue. Thromb. Haemost. 2010, 104, 1124–1132.
- Del Rosso, M.; Margheri, F.; Serratì, S.; Chillà, A.; Laurenzana, A.; Fibbi, G. The urokinase receptor system, a key regulator at the intersection between inflammation, immunity, and coagulation. Curr. Pharm. Des. 2011, 17, 1924–1943.
- Napolitano, F.; Montuori, N. The Role of the Plasminogen Activation System in Angioedema: Novel Insights on the Patho-genesis. J. Clin. Med. 2021, 10, 518.
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24.
- Navaratna, D.; Menicucci, G.; Maestas, J.; Srinivasan, R.; McGuire, P.; Das, A. A peptide inhibitor of the urokinase/urokinase receptor system inhibits alteration of the blood-retinal barrier in diabetes. FASEB J. 2008, 22, 3310–3317.
- Kalbasi, A.P.; Patecki, M.; Tkachuk, S.; Kiyan, Y.; Haller, H.; Dumler, I. Urokinase receptor mediates osteoclastogenesis via M-CSF release from osteoblasts and the c-Fms/PI3K/Akt/NF-κB pathway in osteoclasts. J. Bone Miner. Res. 2015, 30, 379–388.
- Tomogane, K.; Kanno, Y.; Kawashita, E.; Okada, K.; Takeuchi, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. The Absence of Urokinase-type Plasminogen Activator Receptor Plays a Role in the Insulin-independent Glucose Metabolism. J. Cardiovasc. Pharmacol. 2011, 57, 334–339.
- Masucci, M.T.; Minopoli, M.; Di Carluccio, G.; Motti, M.L.; Carriero, M.V. Therapeutic Strategies Targeting Urokinase and Its Receptor in Cancer. Cancers 2022, 14, 498.
- Yepes, M.; Woo, Y.; Martin-Jimenez, C. Plasminogen Activators in Neurovascular and Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4380.
- Kumar, A.A.; Buckley, B.J.; Ranson, M. The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules 2022, 12, 152.
- Ismail, A.; Shaker, B.; Bajou, K. The Plasminogen-Activator Plasmin System in Physiological and Pathophysiological Angio-genesis. Int. J. Mol. Sci. 2021, 23, 337.
- Kanno, Y.; Ishisaki, A.; Kawashita, E.; Kuretake, H.; Ikeda, K.; Matsuo, O. uPA Attenuated LPS-induced Inflammatory Osteoclas-togenesis through the Plasmin/PAR-1/Ca2+/CaMKK/AMPK Axis. Int. J. Biol. Sci. 2016, 12, 63–71.
- Dinesh, P.; Rasool, M. uPA/uPAR signaling in rheumatoid arthritis: Shedding light on its mechanism of action. Pharmacol. Res. 2018, 134, 31–39.
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arter. Thromb. Vasc. Biol. 2017, 37, 1446–1452.
- Alfano, D.; Franco, P.; Stoppelli, M.P. Modulation of Cellular Function by the Urokinase Receptor Signalling: A Mechanistic View. Front. Cell Dev. Biol. 2022, 10, 818616.
- Enocsson, H.; Sjöwall, C.; Wetterö, J. Soluble urokinase plasminogen activator receptor--a valuable biomarker in systemic lupus erythematosus? Clin. Chim. Acta 2015, 444, 234–241.
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943.
- Czekay, R.-P.; Kuemmel, T.A.; Orlando, R.A.; Farquhar, M.G. Direct Binding of Occupied Urokinase Receptor (uPAR) to LDL Receptor-related Protein Is Required for Endocytosis of uPAR and Regulation of Cell Surface Urokinase Activity. Mol. Biol. Cell 2001, 12, 1467–1479.
- Chavakis, T.; Kanse, S.M.; Yutzy, B.; Lijnen, H.R.; Preissner, K.T. Vitronectin concentrates proteolytic activity on the cell surface and extracellular matrix by trapping soluble urokinase receptor-urokinase complexes. Blood 1998, 91, 2305–2312.
- Selleri, C.; Montuori, N.; Ricci, P.; Visconte, V.; Carriero, M.V.; Sidenius, N.; Serio, B.; Blasi, F.; Rotoli, B.; Rossi, G. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood 2005, 105, 2198–2205.
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36.
- Li Santi, A.; Napolitano, F.; Montuori, N.; Ragno, P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 22084111.
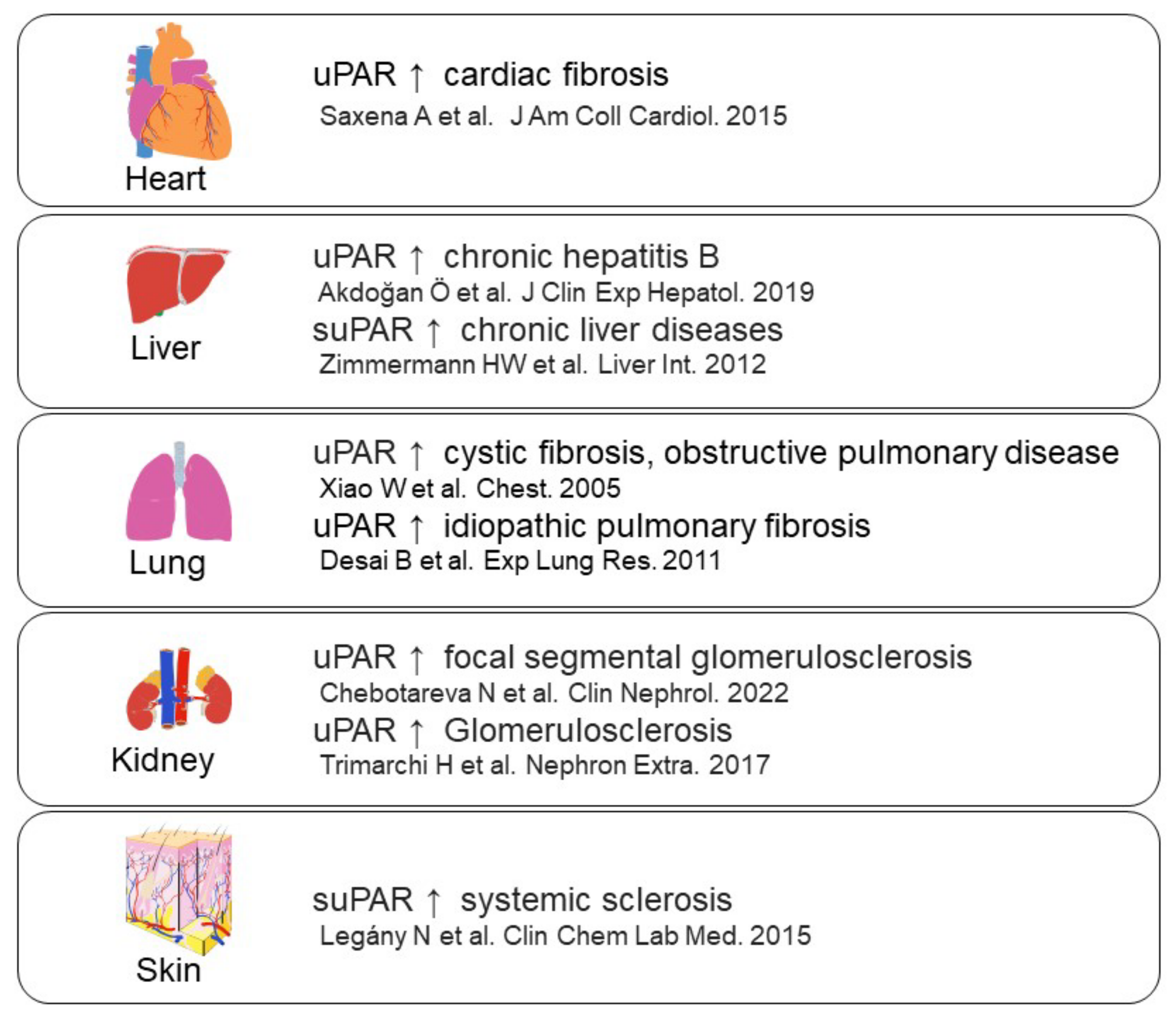
- Saxena, A.; Izmirly, P.M.; Han, S.W.; Briassouli, P.; Rivera, T.L.; Zhong, H.; Friedman, D.M.; Clancy, R.M.; Buyon, J.P. Serum Biomarkers of Inflammation, Fibrosis, and Cardiac Function in Facilitating Diagnosis, Prognosis, and Treatment of Anti-SSA/Ro-Associated Cardiac Neonatal Lupus. J. Am. Coll. Cardiol. 2015, 66, 930–939.
- Akdoğan, J.P.; Yücel, A.A.; Sargin, Z.G.; Sönmez, C.; Yilmaz, G.E.; Özenirler, S. Evaluation of Plasma Urokinase-Type Plasminogen Activator Receptor (UPAR) in Patients with Chronic Hepatitis B, C and Non-Alcoholic Fatty Liver Disease (NAFLD) as Serological Fibrosis Marker. J. Clin. Exp. Hepatol. 2019, 9, 29–33.
- Zimmermann, H.W.; Koch, A.; Seidler, S.; Trautwein, C.; Tacke, F. Circulating soluble urokinase plasminogen activator is elevated in patients with chronic liver disease, discriminates stage and aetiology of cirrhosis and predicts prognosis. Liver Int. 2012, 32, 500–509.
- Xiao, W.; Hsu, Y.-P.; Ishizaka, A.; Kirikae, T.; Moss, R.B. Sputum Cathelicidin, Urokinase Plasminogen Activation System Components, and Cytokines Discriminate Cystic Fibrosis, COPD, and Asthma Inflammation. Chest 2005, 128, 2316–2326.
- Desai, B.; Mattson, J.; Paintal, H.; Nathan, M.; Shen, F.; Beaumont, M.; Malinao, M.C.; Li, Y.; Canfield, J.; Basham, B. Differential expression of monocyte/macrophage-selective markers in human idiopathic pulmonary fibrosis. Exp. Lung Res. 2011, 37, 227–238.
- Chebotareva, N.; Vinogradov, A.; Cao, V.; Gindis, A.; Berns, A.; Alentov, I.; Sergeeva, N. Serum levels of plasminogen activator urokinase receptor and cardiotrophin-like cytokine factor 1 in patients with nephrotic syndrome. Clin. Nephrol. 2022, 97, 103–110.
- Trimarchi, H.; Canzonieri, R.; Schiel, A.; Costales-Collaguazo, C.; Stern, A.; Paulero, M.; Rengel, T.; Andrews, J.; Iotti, A.; Forrester, M. In IgA Nephropathy, Glomerulo-sclerosis Is Associated with Increased Urinary CD80 Excretion and Urokinase-Type Plasminogen Activator Receptor-Positive Podocyturia. Nephron Extra 2017, 7, 52–61.
- Legány, N.; Toldi, G.; Distler, J.H.; Beyer, C.; Szalay, B.; Kovács, L.; Vásárhelyi, B.; Balog, A. Increased plasma soluble urokinase plasminogen activator receptor levels in systemic sclerosis: Possible association with microvascular abnormalities and extent of fibrosis. Clin. Chem. Lab. Med. 2015, 53, 1799–1805.
- Egea-Zorrilla, A.; Vera, L.; Saez, B.; Pardo-Saganta, A. Promises and Challenges of Cell-Based Therapies to Promote Lung Re-generation in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 2595.
- Kanno, Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. Int. J. Mol. Sci. 2019, 20, 619.
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Thromb. Haemost. 2013, 15, 215–219.
- Hasanzadeh, A.; Rafiei, A.; Kazemi, M.; Beiromvand, M.; Bahreini, A.; Khanahmad, H. The Role of Tissue Inhibitor of Metallopro-teinase-1 and 2 in Echinococcus granulosus senso lato-Induced Human Hepatic Fibrosis. Acta Parasitol. 2022, 67, 851–857.
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2019, 16, 11–31.
- Horowitz, J.C.; Tschumperlin, D.J.; Kim, K.K.; Osterholzer, J.J.; Subbotina, N.; Ajayi, I.O.; Teitz-Tennenbaum, S.; Virk, A.; Dotson, M.; Liu, F. Urokinase Plasminogen Activator Overex-pression Reverses Established Lung Fibrosis. Thromb Haemost. 2019, 119, 1968–1980.
- Sun, C.; Li, D.G.; Chen, Y.W.; Chen, Y.W.; Wang, B.C.; Sun, Q.L.; Lu, H.M. Transplantation of urokinase-type plasminogen activator gene-modified bone marrow-derived liver stem cells reduces liver fibrosis in rats. J. Gene Med. 2008, 10, 855–866.
- Dergilev, K.; Beloglazova, I.; Tsokolaeva, Z.; Vasilets, Y.; Parfenova, E. Deficiency of Urokinase-Type Plasminogen Activator Receptor Is Associated with the Development of Perivascular Fibrosis in Mouse Heart. Bull. Exp. Biol. Med. 2022, 173, 5–9.
- Manetti, M.; Rosa, I.; Milia, A.F.; Guiducci, S.; Carmeliet, P.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: A new model of experimental scleroderma? Ann. Rheum. Dis. 2014, 73, 1700–1709.
- Zhang, G.; Kim, H.; Cai, X.; López-Guisa, J.M.; Alpers, C.E.; Liu, Y.; Carmeliet, P.; Eddy, A.A. Urokinase Receptor Deficiency Accelerates Renal Fibrosis in Obstructive Nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1254–1271.
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.K.; Saleem, M. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960.
- Wei, C.; Möller, C.C.; Altintas, M.M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2007, 14, 55–63.
- Wei, C.; Li, J.; Adair, B.D.; Zhu, K.; Cai, J.; Merchant, M.; Samelko, B.; Liao, Z.; Koh, K.H.; Tardi, N.J. uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J. Clin. Investig. 2019, 129, 1946–1959.
- Dal Monte, M.; Cammalleri, M.; Pecci, V.; Carmosino, M.; Procino, G.; Pini, A.; De Rosa, M.; Pavone, V.; Svelto, M.; Bagnoli, P. Inhibiting the urokinase-type plasminogen activator receptor system recovers STZ-induced diabetic nephropathy. J. Cell. Mol. Med. 2019, 23, 1034–1049.
- Schuliga, M.; Grainge, C.; Westall, G.; Knight, D. The fibrogenic actions of the coagulant and plasminogen activation systems in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2018, 97, 108–117.
- Oh, H.; Park, H.E.; Song, M.S.; Kim, H.; Baek, J.-H. The Therapeutic Potential of Anticoagulation in Organ Fibrosis. Front. Med. 2022, 9, 866746.
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Investig. 2010, 120, 1950–1960.
- Kanno, Y.; Hirade, K.; Ishisaki, A.; Nakajima, K.; Suga, H.; Into, T.; Matsushita, K.; Okada, K.; Matsuo, O.; Matsuno, H. Lack of alpha2-antiplasmin improves cutaneous wound healing via over-released vascular endothelial growth factor-induced angiogenesis in wound lesions. J. Thromb. Haemost. 2006, 4, 1602–1610.
- Kanno, Y.; Kawashita, E.; Kokado, A.; Kuretake, H.; Ikeda, K.; Okada, K.; Seishima, M.; Ueshima, S.; Matsuo, O.; Matsuno, H. α2AP mediated myofibroblast formation and the development of renal fibrosis in unilateral ureteral obstruction. Sci. Rep. 2014, 4, srep05967.
- Kanno, Y.; Kuroki, A.; Okada, K.; Tomogane, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. alpha2-Antiplasmin is involved in the production of transforming growth factor beta1 and fibrosis. J. Thromb. Haemost. 2007, 5, 2266–2273.
- Beier, J.I.; Kaiser, J.P.; Guo, L.; Martínez-Maldonado, M.; Arteel, G.E. Plasminogen activator inhibitor-1 deficient mice are protected from angiotensin II-induced fibrosis. Arch. Biochem. Biophys. 2011, 510, 19–26.
- Chuang-Tsai, S.; Sisson, T.H.; Hattori, N.; Tsai, C.G.; Subbotina, N.M.; Hanson, K.E.; Simon, R.H. Reduction in Fibrotic Tissue Formation in Mice Genetically Deficient in Plasminogen Activator Inhibitor. Am. J. Pathol. 2003, 163, 445–452.
- Ortiz-Zapater, E.; Signes-Costa, J.; Montero, P.; Roger, I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts? Biomedicines 2022, 10, 1423.
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules 2021, 11, 1095.
- Wei, J.; Xu, Z.; Yan, X. The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front. Immunol. 2022, 13, 934377.
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945.
- Wang, F.; Xia, H.; Yao, S. Regulatory T cells are a double-edged sword in pulmonary fibrosis. Int. Immunopharmacol. 2020, 84, 106443.
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2015, 7, 8809–8822.
- Derada Troletti, C.; Fontijn, R.D.; Gowing, E.; Charabati, M.; van Het Hof, B.; Didouh, I.; van der Pol, S.M.A.; Geerts, D.; Prat, A.; van Horssen, J. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 2019, 10, 45.
- Wu, N.; Wang, Y.; Wang, K.; Zhong, B.; Liao, Y.; Liang, J.; Jiang, N. Cathepsin K regulates the tumor growth and metastasis by IL-17/CTSK/EMT axis and mediates M2 macrophage polarization in castration-resistant prostate cancer. Cell Death Dis. 2022, 13, 813.
- Paquissi, F.; Abensur, H. The Th17/IL-17 Axis and Kidney Diseases, With Focus on Lupus Nephritis. Front Med. 2021, 8, 654912.
- Zhang, H.; Phan, S. Inhibition of myofibroblast apoptosis by transforming growth factor beta. Am. J. Respir. Cell Mol. Biol. 1999, 21, 658–665.
- Tang, W.W.; Ulich, T.R.; Lacey, D.L.; Hill, D.C.; Qi, M.; Kaufman, S.A.; Van, G.Y.; Tarpley, J.E.; Yee, J.S. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am. J. Pathol. 1996, 148, 1169–1180.
- Iekushi, K.; Taniyama, Y.; Azuma, J.; Sanada, F.; Kusunoki, H.; Yokoi, T.; Koibuchi, N.; Okayama, K.; Rakugi, H.; Morishita, R. Hepatocyte growth factor attenuates renal fibrosis through TGF-β1 suppression by apoptosis of myofibroblasts. J. Hypertens. 2010, 28, 2454–2461.
- Yoshimine, H.; Tanoue, S.; Ibi, Y.; Minami, M.; Nakahara, M.; Tokunaga, K.; Kanmura, S.; Ido, A. Hepatocyte growth factor ameliorates methyl-glyoxal-induced peritoneal inflammation and fibrosis in mouse model. Clin. Exp. Nephrol. 2021, 25, 935–943.
- Jeffers, A.; Qin, W.; Owens, S.; Koenig, K.B.; Komatsu, S.; Giles, F.J.; Schmitt, D.M.; Idell, S.; Tucker, T.A. Glycogen Synthase Kinase-3β Inhibition with 9-ING-41 At-tenuates the Progression of Pulmonary Fibrosis. Sci. Rep. 2019, 9, 18925.
- Kochtebane, N.; Choqueux, C.; Passefort, S.; Nataf, P.; Messika-Zeitoun, D.; Bartagi, A.; Michel, J.B.; Anglés-Cano, E.; Jacob, M.P. Plasmin induces apoptosis of aortic valvular myofibroblasts. J. Pathol. 2009, 221, 37–48.
- He, Y.; Tsou, P.; Khanna, D.; Sawalha, A. Methyl-CpG-binding protein 2 mediates antifibrotic effects in scleroderma fibroblasts. Ann. Rheum. Dis. 2018, 77, 1208–1218.
- Sugioka, K.; Nishida, T.; Kodama-Takahashi, A.; Murakami, J.; Mano, F.; Okada, K.; Fukuda, M.; Kusaka, S. Urokinase-type plasminogen activator negatively regulates α-smooth muscle actin expression via Endo180 and the uPA receptor in corneal fibroblasts. Am. J. Physiol. Physiol. 2022, 323, C104–C115.
- Wang, L.; Ly, C.M.; Ko, C.-Y.; Meyers, E.E.; Lawrence, D.A.; Bernstein, A.M. uPA Binding to PAI-1 Induces Corneal Myofibroblast Differentiation on Vitronectin. Investig. Opthalmology Vis. Sci. 2012, 53, 4765–4775.
- Vorstandlechner, V.; Laggner, M.; Copic, D.; Klas, K.; Direder, M.; Chen, Y.; Golabi, B.; Haslik, W.; Radtke, C.; Tschachler, E. The serine proteases dipeptidyl-peptidase 4 and urokinase are key molecules in human and mouse scar formation. Nat. Commun. 2021, 12, 6242.
- Bernstein, A.M.; Twining, S.S.; Warejcka, D.J.; Tall, E.; Masur, S.K. Urokinase Receptor Cleavage: A Crucial Step in Fibroblast-to-Myofibroblast Differentiation. Mol. Biol. Cell 2007, 18, 2716–2727.
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934.
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286.
- Wang, P.; Ma, M.; Zhang, S. EGF-induced urokinase plasminogen activator receptor promotes epithelial to mesenchymal tran-sition in human gastric cancer cells. Oncol. Rep. 2017, 38, 2325–2334.
- Laurenzana, A.; Biagioni, A.; Bianchini, F.; Peppicelli, S.; Chillà, A.; Margheri, F.; Luciani, C.; Pimpinelli, N.; Del Rosso, M.; Calorini, L. Inhibition of uPAR-TGFβ crosstalk blocks MSC-dependent EMT in melanoma cells. J. Mol. Med. 2015, 93, 783–794.
- Wang, Q.; Wang, Y.; Zhang, Y.; Zhang, Y.; Xiao, W. The role of uPAR in epithelial-mesenchymal transition in small airway epi-thelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013, 14, 67.
- Lester, R.D.; Jo, M.; Montel, V.; Takimoto, S.; Gonias, S.L. uPAR induces epithelial–mesenchymal transition in hypoxic breast cancer cells. J. Cell Biol. 2007, 178, 425–436.
- Hannan, R.T.; Miller, A.E.; Hung, R.-C.; Sano, C.; Peirce, S.M.; Barker, T.H. Extracellular matrix remodeling associated with bleomycin-induced lung injury supports pericyte-to-myofibroblast transition. Matrix Biol. Plus 2020, 10, 100056.
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C Induces Phenotypic Changes in Fibroblasts to Myofibroblasts with High Contractility through the Integrin αvβ1/Transforming Growth Factor β/SMAD Signaling Axis in Human Breast Cancer. Am. J. Pathol. 2020, 190, 2123–2135.
- Bianchini, F.; Peppicelli, S.; Fabbrizzi, P.; Biagioni, A.; Mazzanti, B.; Menchi, G.; Calorini, L.; Pupi, A.; Trabocchi, A. Triazole RGD antagonist reverts TGFβ1-induced endothelial-to-mesenchymal transition in endothelial precursor cells. Mol. Cell. Biochem. 2016, 424, 99–110.
- Borok, Z. Role for α3 integrin in EMT and pulmonary fibrosis. J. Clin. Investig. 2008, 119, 7–10.
- Shochet, G.E.; Brook, E.; Bardenstein-Wald, B.; Grobe, H.; Edelstein, E.; Israeli-Shani, L.; Shitrit, D. Integrin alpha-5 silencing leads to my-ofibroblastic differentiation in IPF-derived human lung fibroblasts. Ther. Adv. Chronic Dis. 2020, 24, 2040622320936023.
- Overstreet, J.M.; Wang, Y.; Wang, X.; Niu, A.; Gewin, L.S.; Yao, B.; Harris, R.C.; Zhang, M.Z. Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. FASEB J. 2017, 31, 4407–4421.
- Xu, H.; Liu, L.; Cong, M.; Liu, T.; Sun, S.; Ma, H.; You, H.; Jia, J.; Wang, P. EGF neutralization antibodies attenuate liver fibrosis by inhibiting myofi-broblast proliferation in bile duct ligation mice. Histochem. Cell Biol. 2020, 154, 107–116.
- Shu, D.Y.; Lovicu, F.J. Enhanced EGF receptor-signaling potentiates TGFβ-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 185, 107693.
- Wu, S.; Yang, S.; Qu, H. circ_CHFR regulates ox-LDL-mediated cell proliferation, apoptosis, and EndoMT by miR-15a-5p/EGFR axis in human brain microvessel endothelial cells. Open Life Sci. 2021, 16, 1053–1063.
- Thooyamani, A.S.; Mukhopadhyay, A. PDGFRα mediated survival of myofibroblasts inhibit satellite cell proliferation during aberrant regeneration of lacerated skeletal muscle. Sci. Rep. 2021, 11, 1–15.
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570.
- Yan, D.; Liu, X.; Xu, H.; Guo, S.-W. Platelets induce endothelial–mesenchymal transition and subsequent fibrogenesis in endometriosis. Reprod. Biomed. Online 2020, 41, 500–517.
- Chen, L.; Lin, G.; Chen, K.; Liang, R.; Wan, F.; Zhang, C.; Tian, G.; Zhu, X. VEGF promotes migration and invasion by regulating EMT and MMPs in nasopharyngeal carcinoma. J. Cancer 2020, 11, 7291–7301.
- Modi, S.; Tiwari, A.; Kulkarni, V. Reversal of TGF-β-induced epithelial-mesenchymal transition in hepatocellular carcinoma by sorafenib, a VEGFR-2 and Raf kinase inhibitor. Curr. Res. Pharmacol. Drug. Discov. 2021, 2, 100014.
- Rossato, F.A.; Su, Y.; Mackey, A.; Ng, Y.S.E. Fibrotic Changes and Endothelial-to-Mesenchymal Transition Promoted by VEGFR2 Antagonism Alter the Therapeutic Effects of VEGFA Pathway Blockage in a Mouse Model of Choroidal Neovascularization. Cells 2020, 9, 2057.
- Sanders, Y.Y.; Cui, Z.; Le Saux, C.J.; Horowitz, J.C.; Rangarajan, S.; Kurundkar, A.; Antony, V.B.; Thannickal, V.J. SMAD-Independent Down-Regulation of Caveolin-1 by TGF-β: Effects on Proliferation and Survival of Myofibroblasts. PLoS ONE 2015, 10, e0116995.
- Yang, S.; Zhao, J.; Huang, S.; Shu, B.; Yang, R.; Chen, L.; Xu, Y.; Xie, J.; Liu, X.; Jia, J. Reduced hydration-induced decreased caveolin-1 expression causes epithelial-to-mesenchymal transition. Am. J. Transl. Res. 2020, 12, 8067–8083.
- Jung, A.C.; Ray, A.M.; Ramolu, L.; Macabre, C.; Simon, F.; Noulet, F.; Blandin, A.F.; Renner, G.; Lehmann, M.; Choulier, L. Caveolin-1-negative head and neck squamous cell carcinoma primary tumors display increased epithelial to mesenchymal transition and prometastatic properties. Oncotarget 2015, 6, 41884–41901.
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 Deficiency Induces Spontaneous Endothelial-to-Mesenchymal Transition in Murine Pulmonary Endothelial Cells in Vitro. Am. J. Pathol. 2012, 182, 325–331.
- Schnieder, J.; Mamazhakypov, A.; Birnhuber, A.; Wilhelm, J.; Kwapiszewska, G.; Ruppert, C.; Markart, P.; Wujak, L.; Rubio, K.; Barreto, G. Loss of LRP1 promotes acquisition of contractile-myofibroblast phenotype and release of active TGF-β1 from ECM stores. Matrix Biol. 2019, 88, 69–88.
- Hu, K.; Lin, L.; Tan, X.; Yang, J.; Bu, G.; Mars, W.M.; Liu, Y. tPA Protects Renal Interstitial Fibroblasts and Myofibroblasts from Apoptosis. J. Am. Soc. Nephrol. 2008, 19, 503–514.
- Hu, K.; Wu, C.; Mars, W.M.; Liu, Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor–related protein 1–mediated integrin signaling. J. Clin. Investig. 2007, 117, 3821–3832.
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of Plasminogen Activator Inhibitor-1 Attenuates Transforming Growth Factor-β-Dependent Epithelial Mesenchymal Transition and Differentiation of Fibroblasts to Myofibroblasts. PLoS ONE 2016, 11, e0148969.
- Zhang, Y.P.; Wang, W.L.; Liu, J.; Li, W.B.; Bai, L.L.; Yuan, Y.D.; Song, S.X. Plasminogen activator inhibitor-1 promotes the proliferation and inhibits the apoptosis of pulmonary fibroblasts by Ca2+ signaling. Thromb. Res. 2012, 131, 64–71.
- Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; Miyata, Y. Inhibition of PAI-1 limits chemotherapy resistance in lung cancer through suppressing myofibroblast characteristics of cancer-associated fibroblasts. J. Cell. Mol. Med. 2019, 23, 2984–2994.
- Pedroja, B.; Kang, L.; Imas, A.; Carmeliet, P.; Bernstein, A. Plasminogen activator inhibitor-1 regulates integrin alphavbeta3 ex-pression and autocrine transforming growth factor beta signaling. J. Biol. Chem. 2009, 284, 20708–20717.
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507.
- Ghosh, A.K.; Bradham, W.S.; Gleaves, L.A.; De Taeye, B.; Murphy, S.B.; Covington, J.W.; Vaughan, D.E. Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: Involvement of constitutive transforming growth factor-beta signaling and endothelial-to-mesenchymal transition. Circulation 2010, 122, 1200–1209.
- Baumeier, C.; Escher, F.; Aleshcheva, G.; Pietsch, H.; Schultheiss, H.-P. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res. Cardiol. 2021, 116, 1–9.
- Kanno, Y.; Kawashita, E.; Kokado, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-antiplasmin regulates the development of dermal fibrosis in mice by prostaglandin F2α synthesis through adipose triglyceride lipase/calcium-independent phospho-lipase. Arthritis Rheum. 2013, 65, 492–502.
- Kanno, Y.; Shu, E.; Kanoh, H.; Seishima, M. The Antifibrotic Effect of α2AP Neutralization in Systemic Sclerosis Dermal Fibroblasts and Mouse Models of Systemic Sclerosis. J. Investig. Dermatol. 2015, 136, 762–769.
- Kanno, Y.; Shu, E.; Niwa, H.; Seishima, M.; Ozaki, K.-I. MicroRNA-30c attenuates fibrosis progression and vascular dysfunction in systemic sclerosis model mice. Mol. Biol. Rep. 2021, 48, 3431–3437.
- Kanno, Y.; Hirota, M.; Matsuo, O.; Ozaki, K.-I. α2-antiplasmin positively regulates endothelial-to-mesenchymal transition and fibrosis progression in diabetic nephropathy. Mol. Biol. Rep. 2021, 49, 205–215.
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New insights into fibrosis from the ECM degradation perspective: The macro-phage-MMP-ECM interaction. Cell Biosci. 2022, 12, 117.
- Michalczyk, K.; Cymbaluk-Płoska, A. Metalloproteinases in Endometrial Cancer-Are They Worth Measuring? Int. J. Mol. Sci. 2021, 22, 12472.
- Niwa, H.; Kanno, Y.; Shu, E.; Seishima, M. Decrease in matrix metalloproteinase-3 activity in systemic sclerosis fibroblasts causes α2-antiplasmin and extracellular matrix deposition, and contributes to fibrosis development. Mol. Med. Rep. 2020, 22, 3001–3007.
- Lijnen, H.R. Matrix Metalloproteinases and Cellular Fibrinolytic Activity. Biochemistry 2002, 67, 92–98.
- Menou, A.; Duitman, J.; Crestani, B. The impaired proteases and anti-proteases balance in Idiopathic Pulmonary Fibrosis. Matrix Biol. 2018, 68-69, 382–403.
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337.
- Okazaki, I.; Noro, T.; Tsutsui, N.; Yamanouchi, E.; Kuroda, H.; Nakano, M.; Yokomori, H.; Inagaki, Y. Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs). Cancers 2014, 6, 1220–1255.
- Waszczykowska, A.; Podgórski, M.; Waszczykowski, M.; Gerlicz-Kowalczuk, Z.; Jurowski, P. Matrix Metalloproteinases MMP-2 and MMP-9, Their Inhibitors TIMP-1 and TIMP-2, Vascular Endothelial Growth Factor and sVEGFR-2 as Predictive Markers of Ischemic Retinopathy in Patients with Systemic Sclerosis-Case Series Report. Int. J. Mol. Sci. 2020, 21, 8703.
- Young-Min, A.S.; Beeton, C.; Laughton, R.; Plumpton, T.; Bartram, S.; Murphy, G.; Black, C.; Cawston, E.T. Serum TIMP-1, TIMP-2, and MMP-1 in patients with systemic sclerosis, primary Raynaud’s phenomenon, and in normal controls. Ann. Rheum. Dis. 2001, 60, 846–851.
- Yao, H.; Yang, X.; Yan, M.; Fang, X.; Wang, Y.; Qi, H.; Sun, L. Correlation of Serum M-CSF, CER, and TIMP-1 Levels with Liver Fibrosis in Viral Hepatitis. Comput. Math. Methods Med. 2022, 2022, 6736225.
- Eguchi, A.; Iwasa, M.; Sugimoto, R.; Tempaku, M.; Yoshikawa, K.; Yoshizawa, N.; Povero, D.; Sugimoto, K.; Hasegawa, H.; Takei, Y. Complement complex 1 subunit q-mediated hepatic stellate cell activation with connective tissue growth factor elevation is a prognostic factor for survival in rat and human chronic liver diseases. Hepatol. Commun. 2022, 6, 3515–3527.
- Lefeuvre, C.; Roux, M.; Blanchard, S.; Le Guillou-Guillemette, H.; Boursier, J.; Lunel-Fabiani, F.; Jeannin, P.; Pivert, A.; Ducancelle, A. Analysis of hepatic fibrosis markers in the serum of chronic hepatitis B patients according to basal core promoter/precore mutants. Sci. Rep. 2022, 12, 10261.
- Yang, K.; Palm, J.; König, J.; Seeland, U.; Rosenkranz, S.; Feiden, W.; Rübe, C.; Rübe, C.E. Matrix-Metallo-Proteinases and their tissue inhibitors in radiation-induced lung injury. Int. J. Radiat. Biol. 2007, 83, 665–676.
- Wang, Y.; Huang, G.; Mo, B.; Wang, C. Artesunate modulates expression of matrix metalloproteinases and their inhibitors as well as collagen-IV to attenuate pulmonary fibrosis in rats. Genet. Mol. Res. 2016, 15, 530.
- Kökény, G.; Németh, Á.; Kopp, J.B.; Chen, W.; Oler, A.J.; Manzéger, A.; Rosivall, L.; Mózes, M.M. Susceptibility to kidney fibrosis in mice is associated with early growth response-2 protein and tissue inhibitor of metalloproteinase-1 expression. Kidney Int. 2022, 102, 337–354.
- Takawale, A.; Zhang, P.; Patel, V.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myo-cardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103.
- Kanno, Y.; Shu, E.; Niwa, H.; Kanoh, H.; Seishima, M. Alternatively activated macrophages are associated with the α2AP production that occurs with the development of dermal fibrosis: The role of alternatively activated macrophages on the development of fibrosis. Arthritis Res Ther. 2020, 22, 76.
- Kanno, Y.; Kawashita, E.; Minamida, M.; Kaneiwa, A.; Okada, K.; Ueshima, S.; Matsuo, O.; Matsuno, H. Alpha2-antiplasmin is associated with the pro-gression of fibrosis. Am. J. Pathol. 2010, 176, 238–245.
- Kanno, Y.; Miyashita, M.; Seishima, M.; Matsuo, O. α2AP is associated with the development of lupus nephritis through the regulation of plasmin inhibition and inflammatory responses. Immun. Inflamm. Dis. 2020, 8, 267–278.
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen Activator Inhibitor Type-1 as a Regulator of Fibrosis. J. Cell Biochem. 2018, 119, 17–27.
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L740–L752.
- Ma, L.; Fogo, A. PAI-1 and kidney fibrosis. Front. Biosci. 2009, 14, 2028–2041.
- Liakouli, V.; Cipriani, P.; Marrelli, A.; Alvaro, S.; Ruscitti, P.; Giacomelli, R. Angiogenic cytokines and growth factors in systemic sclerosis. Autoimmun. Rev. 2011, 10, 590–594.
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786.
- Zanin-Silva, D.C.; Santana-Gonçalves, M.; Kawashima-Vasconcelos, M.Y.; Oliveira, M.C. Management of Endothelial Dysfunction in Systemic Sclerosis: Current and Developing Strategies. Front. Med. 2021, 8, 250.
- Plow, E.F.; Hoover-Plow, J. The Functions of Plasminogen in Cardiovascular Disease. Trends Cardiovasc. Med. 2004, 14, 180–186.
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904.
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285.
- Yan, Q.; Sage, E. Transforming growth factor-beta1 induces apoptotic cell death in cultured retinal endothelial cells but not pericytes: Association with decreased expression of p21waf1/cip. J. Cell Biochem. 1998, 70, 70–83.
- Long, D.; Yang, J.; Wu, X.; Gui, Y.; Yu, L. Urokinase-type plasminogen activator protects human umbilical vein endothelial cells from apoptosis in sepsis. Int. J. Clin. Exp. Pathol. 2019, 12, 77–86.
- Prager, G.W.; Mihaly, J.; Brunner, P.M.; Koshelnick, Y.; Hoyer-Hansen, G.; Binder, B.R. Urokinase mediates endothelial cell survival via induction of the X-linked inhibitor of apoptosis protein. Blood 2009, 113, 1383–1390.
- Song, T.; Meng, S.; Xu, S.-T.; Jin, S.-J.; Zeng, Q.-Z.; Gu, G.-J. The overexpression of uPA promotes the proliferation and fibrinolytic activity of human umbilical vein endothelial cells. Int. J. Clin. Exp. Pathol. 2019, 12, 2959–2966.
- Beloglazova, I.B.; Zubkova, E.S.; Stambol’skii, D.V.; Plekhanova, O.S.; Men’shikov, M.Y.; Akopyan, Z.A.; Bibilashvili, R.S.; Parfenova, E.V.; Tkachuk, V.A. Proteolytically inactive recom-binant forms of urokinase suppress migration of endothelial cells. Bull. Exp. Biol. Med. 2014, 156, 756–759.
- Balsara, R.D.; Merryman, R.; Virjee, F.; Northway, C.; Castellino, F.J.; Ploplis, A.V. A deficiency of uPAR alters endothelial angiogenic function and cell morphology. Vasc. Cell 2011, 3, 10.
- Lu, H.; Mabilat, C.; Yeh, P.; Guitton, J.D.; Li, H.; Pouchelet, M.; Shoevaert, D.; Legrand, Y.; Soria, J.; Soria, C. Blockage of urokinase receptor reduces in vitro the motility and the deformability of endothelial cells. FEBS Lett. 1996, 380, 21–24.
- Beloglazova, I.; Stepanova, V.; Zubkova, E.; Dergilev, K.; Koptelova, N.; Tyurin-Kuzmin, P.A.; Dyikanov, D.; Plekhanova, O.; Cines, D.B.; Mazar, A.P. Mesenchymal stromal cells enhance self-assembly of a HUVEC tubular network through uPA-uPAR/VEGFR2/integrin/NOTCH crosstalk. Biochim. Biophys. Acta. Mol. Cell Res. 2022, 1869, 119157.
- Alexander, R.A.; Prager, G.W.; Mihaly-Bison, J.; Uhrin, P.; Sunzenauer, S.; Binder, B.R.; Schütz, G.J.; Freissmuth, M.; Breuss, J.M. VEGF-induced endothelial cell migration requires urokinase receptor (uPAR)-dependent integrin redistribution. Cardiovasc. Res. 2012, 94, 125–135.
- Reuning, U.; Sperl, S.; Kopitz, C.; Kessler, H.; Krüger, A.; Schmitt, M.; Magdolen, V. Urokinase-type Plasminogen Activator (uPA) and its Receptor (uPAR): Development of Antagonists of uPA / uPAR Interaction and their Effects In Vitro and In Vivo. Curr. Pharm. Des. 2003, 9, 1529–1543.
- Poettler, M.; Unseld, M.; Mihaly-Bison, J.; Uhrin, P.; Koban, F.; Binder, B.R.; Zielinski, C.C.; Prager, G.W. The urokinase receptor (CD87) represents a central mediator of growth factor-induced endothelial cell migration. Thromb. Haemost. 2012, 108, 357–366.
- Stepanova, V.; Jayaraman, P.S.; Zaitsev, S.V.; Lebedeva, T.; Bdeir, K.; Kershaw, R.; Holman, K.R.; Parfyonova, Y.V.; Semina, E.V.; Beloglazova, I.B. Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodomain Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J. Biol. Chem. 2016, 291, 15029–15045.
- Raghu, H.; Nalla, A.K.; Gondi, C.S.; Gujrati, M.; Dinh, D.H.; Rao, J.S. uPA and uPAR shRNA inhibit angiogenesis via enhanced secretion of SVEGFR1 independent of GM-CSF but dependent on TIMP-1 in endothelial and glioblastoma cells. Mol. Oncol. 2011, 6, 33–47.
- Park, J.; Keller, G.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subep-ithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell. 1993, 4, 1317–1326.
- Kanno, Y.; Shu, E.; Kanoh, H.; Matsuda, A.; Seishima, M. α2AP regulates vascular alteration by inhibiting VEGF signaling in systemic sclerosis: The roles of α2AP in vascular dysfunction in systemic sclerosis. Arthritis Res. Ther. 2017, 19, 1–8.
- Herkenne, S.; Paques, C.; Nivelles, O.; Lion, M.; Bajou, K.; Pollenus, T.; Fontaine, M.; Carmeliet, P.; Martial, J.A.; Nguyen, N.Q. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci. Signal. 2015, 8, ra117.
- Larusch, G.A.; Merkulova, A.; Mahdi, F.; Shariat-Madar, Z.; Sitrin, R.G.; Cines, D.B.; Schmaier, A.H. Domain 2 of uPAR regulates single-chain uro-kinase-mediated angiogenesis through β1-integrin and VEGFR. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H305–H320.
- LaRusch, G.A.; Mahdi, F.; Shariat-Madar, Z.; Adams, G.; Sitrin, R.G.; Zhang, W.M.; McCrae, K.R.; Schmaier, A.H. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood 2010, 115, 5111–5120.
- D’Alessio, S.; Fibbi, G.; Cinelli, M.; Guiducci, S.; Del Rosso, A.; Margheri, F.; Serratì, S.; Pucci, M.; Kahaleh, B.; Fan, P. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004, 50, 3275–3285.
- Bifulco, K.; Longanesi-Cattani, I.; Gala, M.; DICarluccio, G.; Masucci, M.T.; Pavone, V.; Lista, L.; Arra, C.; Stoppelli, M.P.; Carriero, M.V. The soluble form of urokinase receptor promotes angiogenesis through its Ser⁸⁸-Arg-Ser-Arg-Tyr⁹² chemotactic sequence. J. Thromb. Haemost. 2010, 8, 2789–2799.
- Fuchs, P.Ö.; Calitz, C.; Pavlović, N.; Binet, F.; Solbak, S.M.Ø.; Danielson, U.H.; Kreuger, J.; Heindryckx, F.; Gerwins, P. Fibrin fragment E potentiates TGF-β-induced myofibroblast activation and recruitment. Cell Signal. 2020, 72, 109661.
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 6553.
- Liu, S.-F.; Veetil, N.N.; Li, Q.; Kucherenko, M.M.; Knosalla, C.; Kuebler, W.M. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front. Immunol. 2022, 13, 209.
- Christou, H.; Khalil, R.A. Mechanisms of pulmonary vascular dysfunction in pulmonary hypertension and implications for novel therapies. Am. J. Physiol. Circ. Physiol. 2022, 322, H702–H724.
- Ban, C.; Wang, T.; Zhang, S.; Xin, P.; Liang, L.; Wang, C.; Dai, H. Fibrinolytic system related to pulmonary arterial pressure and lung function of patients with idiopathic pulmonary fibrosis. Clin. Respir. J. 2015, 11, 640–647.
- Levi, M.; Moons, L.; Bouché, A.; Shapiro, S.D.; Collen, D.; Carmeliet, P. Deficiency of Urokinase-Type Plasminogen Activator–Mediated Plasmin Generation Impairs Vascular Remodeling During Hypoxia-Induced Pulmonary Hypertension in Mice. Circulation 2001, 103, 2014–2020.
- Manetti, M.; Allanore, Y.; Revillod, L.; Fatini, C.; Guiducci, S.; Cuomo, G.; Bonino, C.; Riccieri, V.; Bazzichi, L.; Liakouli, V. A genetic variation located in the promoter region of the UPAR (CD87) gene is associated with the vascular complications of systemic sclerosis. Arthritis Rheum. 2010, 63, 247–256.
- Makarova, A.M.; Lebedeva, T.V.; Nassar, T.; Higazi, A.A.; Xue, J.; Carinato, M.E.; Bdeir, K.; Cines, D.B.; Stepanova, V. Urokinase-type Plasminogen Activator (uPA) Induces Pulmonary Microvascular Endothelial Permeability through Low Density Lipoprotein Receptor-related Protein (LRP)-dependent Activation of Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2011, 286, 23044–23053.
- Boccella, S.; Panza, E.; Lista, L.; Belardo, C.; Ianaro, A.; De Rosa, M.; de Novellis, V.; Pavone, V. Preclinical evaluation of the urokinase receptor-derived peptide UPARANT as an anti-inflammatory drug. Inflamm. Res. 2017, 66, 701–709.
- Yoon, S.Y.; Lee, Y.J.; Seo, J.H.; Sung, H.J.; Park, K.H.; Choi, I.K.; Kim, S.J.; Oh, S.C.; Choi, C.W.; Kim, B.S. uPAR expression under hypoxic conditions depends on iNOS modulated ERK phosphorylation in the MDA-MB-231 breast carcinoma cell line. Cell Res. 2006, 16, 75–81.
- Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2020, 21, 5203.
- Abdul, S.; Leebeek, F.; Rijken, D.; Uitte de Willige, S. Natural heterogeneity of α2-antiplasmin: Functional and clinical conse-quences. Blood 2016, 127, 538–545.
- Van Geffen, C.; Deißler, A.; Quante, M.; Renz, H.; Hartl, D.; Kolahian, S. Regulatory Immune Cells in Idiopathic Pulmonary Fibrosis: Friends or Foes? Front. Immunol. 2021, 22, 663203.
- Brown, M.; O’Reilly, S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin. Exp. Immunol. 2018, 195, 310–321.
- Pattanaik, D.; Brown, M.; Postlethwaite, B.; Postlethwaite, A. Pathogenesis of Systemic Sclerosis. Front Immunol. 2015, 6, 272.
- Numajiri, H.; Kuzumi, A.; Fukasawa, T.; Ebata, S.; Yoshizaki-Ogawa, A.; Asano, Y.; Kazoe, Y.; Mawatari, K.; Kitamori, T.; Yoshizaki, A. B Cell Depletion Inhibits Fibrosis via Suppression of Profibrotic Macrophage Differentiation in a Mouse Model of Systemic Sclerosis. Arthritis Rheumatol. 2021, 73, 2086–2095.
- Nakayama, W.; Jinnin, M.; Makino, K.; Kajihara, I.; Makino, T.; Fukushima, S.; Inoue, Y.; Ihn, H. Serum levels of soluble CD163 in patients with systemic sclerosis. Rheumatol. Int. 2010, 32, 403–407.
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 5838.
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969.
- Mondino, A.; Blasi, F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004, 25, 450–455.
- Syrovets, T.; Lunov, O.; Simmet, T. Plasmin as a proinflammatory cell activator. J. Leukoc. Biol. 2012, 92, 509–519.
- Medcalf, R.; Keragala, C. Fibrinolysis: A Primordial System Linked to the Immune Response. Int. J. Mol. Sci. 2021, 22, 3406.
- Hastings, S.; Myles, P.; Medcalf, R. Plasmin, Immunity, and Surgical Site Infection. J. Clin. Med. 2021, 10, 2070.
- Vago, J.P.; Sugimoto, M.A.; Lima, K.M.; Negreiros-Lima, G.L.; Baik, N.; Teixeira, M.M.; Perretti, M.; Parmer, R.J.; Miles, L.A.; Sousa, L.P. Plasminogen and the Plasminogen Receptor, Plg-RKT, Regulate Macrophage Phenotypic, and Functional Changes. Front. Immunol. 2019, 10, 1458.
- Shimazu, H.; Munakata, S.; Tashiro, Y.; Salama, Y.; Dhahri, D.; Eiamboonsert, S.; Ota, Y.; Onoda, H.; Tsuda, Y.; Okada, Y. Pharmacological targeting of plasmin prevents lethality in a murine model of macrophage activation syndrome. Blood 2017, 130, 59–72.
- Li, X.; Syrovets, T.; Genze, F.; Pitterle, K.; Oberhuber, A.; Orend, K.H.; Simmet, T. Plasmin Triggers Chemotaxis of Monocyte-Derived Dendritic Cells Through an Akt2-Dependent Pathway and Promotes a T-Helper Type-1 Response. Arter. Thromb. Vasc. Biol. 2010, 30, 582–590.
- Bryer, S.C.; Fantuzzi, G.; Van Rooijen, N.; Koh, T.J. Urokinase-Type Plasminogen Activator Plays Essential Roles in Macrophage Chemotaxis and Skeletal Muscle Regeneration. J. Immunol. 2008, 180, 1179–1188.
- Meznarich, J.; Malchodi, L.; Helterline, D.; Ramsey, S.A.; Bertko, K.; Plummer, T.; Plawman, A.; Gold, E.; Stempien-Otero, A. Urokinase Plasminogen Activator Induces Pro-Fibrotic/M2 Phenotype in Murine Cardiac Macrophages. PLoS ONE 2013, 8, e57837.
- Kanno, Y.; Ishisaki, A.; Miyashita, M.; Matsuo, O. The blocking of uPAR suppresses lipopolysaccharide-induced inflammatory osteoclastogenesis and the resultant bone loss through attenuation of integrin β3/Akt pathway. Immun. Inflamm. Dis. 2016, 4, 338–349.
- Liu, G.; Yang, Y.; Yang, S.; Banerjee, S.; De Freitas, A.; Friggeri, A.; Davis, K.I.; Abraham, E. The receptor for urokinase regulates TLR2 mediated in-flammatory responses in neutrophils. PLoS ONE 2011, 6, e25843.
- Li, J.; Pan, Y.; Li, D.; Xia, X.; Jiang, Q.; Dou, H.; Hou, Y. Urokinase-type plasminogen activator receptor is required for impairing toll-like receptor 7 signaling on macrophage efferocytosis in lupus. Mol. Immunol. 2020, 127, 38–45.
- Kiyan, Y.; Tkachuk, S.; Rong, S.; Gorrasi, A.; Ragno, P.; Dumler, I.; Haller, H.; Shushakova, N. TLR4 Response to LPS Is Reinforced by Urokinase Receptor. Front. Immunol. 2020, 11, 3550.
- Rasmussen, L.J.H.; Petersen, J.E.V.; Eugen-Olsen, J. Soluble Urokinase Plasminogen Activator Receptor (suPAR) as a Biomarker of Systemic Chronic Inflammation. Front. Immunol. 2021, 12, 641.
- Yousif, A.M.; Minopoli, M.; Bifulco, K.; Ingangi, V.; Di Carluccio, G.; Merlino, F.; Motti, M.L.; Grieco, P.; Carriero, M.V. Cyclization of the Urokinase Receptor-Derived Ser-Arg-Ser-Arg-Tyr Peptide Generates a Potent Inhibitor of Trans-Endothelial Migration of Monocytes. PLoS ONE 2015, 10, e0126172.